


Publié le 05 oct 2010Lecture 12 min
Recommandations sur l'hypertension artérielle pulmonaire
F. DELAHAYE, Lyon
Les conclusions de la société européenne de cardiologie sur le diagnostic et le traitement de l’hypertension pulmonaire.
Les Sociétés européennes de cardiologie et de pneumologie ont récemment publié des recommandations[1] sur le diagnostique et le traitement de l’HTAP, qui ont été endossées par la Société française de cardiologie. Les classes des recommandations, les niveaux de preuve sont bien connus.
Classification de l’hypertension pulmonaire
L’HTP est une condition hémodynamique et physiopathologique définie comme une augmentation de la PAP moyenne ≥ 25 mmHg au repos, mesurée par cathétérisme des cavités cardiaques droites (tableau 1).
Elle peut être vue dans de nombreuses conditions cliniques, classées dans 6 groupes selon leurs caractéristiques anatomopathologiques, physiopathologiques et thérapeutiques (tableau 2). Malgré des élévations comparables de la PAP et des résistances vasculaires pulmonaires dans les différents groupes cliniques, les mécanismes, l’approche diagnostique et les implications pronostiques et thérapeutiques sont complètement différents.
La classification des cardiopathies congénitales causant une HTAP peut être clinique (tableau 3) ou anatomo-physiopathologique (tableau 4). L’HTAP « associée » (à une maladie) a une présentation clinique similaire à celle de l’HTAP idiopathique, et une histologie identique, incluant le développement de lésions plexiformes.
La définition de l’HTP à l’effort comme une PAP moyenne > 30 mmHg, mesurée lors d’un cathétérisme des cavités cardiaques droites, n’est pas étayée par des données publiées.
L’HTAP (groupe 1) est une condition clinique caractérisée par la présence d’une HTP précapillaire (tableau 1) en l’absence d’autres causes d’HTP précapillaire comme l’HTP due à une maladie pulmonaire, l’HTP thromboembolique chronique, ou d’autres maladies rares (tableau 2). L’HTAP inclut diverses formes qui partagent un tableau clinique similaire et des modifications anatomopathologiques virtuellement identiques de la microcirculation pulmonaire (tableau 2).
Anatomopathologie de l’hypertension pulmonaire
Les éléments anatomopathologiques sont différents dans les divers groupes cliniques.
• Groupe 1 : les lésions affectent les artères pulmonaires distales (< 500 μm de diamètre) en particulier ; elles sont caractérisées par une hypertrophie de la média, des modifications prolifératives et fibrotiques de l’intima, un épaississement de l’adventice, des lésions complexes (lésions plexiformes, dilatées) et des lésions thrombotiques. Les veines pulmonaires sont habituellement indemnes.
• Groupe 1 : la maladie veinoocclusive pulmonaire atteint les veines septales et les veinules préseptales, avec des lésions fibrotiques occlusives, une muscularisation veineuse, une prolifération capillaire fréquente, un oedème pulmonaire, des hémorragies alvéolaires occultes, une dilatation lymphatique, un grossissement des noeuds lymphatiques et des infiltrats inflammatoires. Les artères pulmonaires distales sont affectées par une hypertrophie médiale, une fibrose intimale et de rares lésions complexes.
• Groupe 2 : les modifications anatomopathologiques sont un élargissement et un épaississement des veines pulmonaires, une dilatation des capillaires pulmonaires, un oedème interstitiel, des hémorragies alvéolaires, et un agrandissement des vaisseaux et des noeuds lymphatiques.
Les artères pulmonaires distales peuvent être affectées par une hypertrophie médiale et une fibrose intimale.
• Groupe 3 : il y a une hypertrophie médiale et une prolifération obstructive de l’intima des artères pulmonaires distales.
• Groupe 4 : des thrombus organisés sont très attachés à la couche médiale des artères pulmonaires, dans les artères pulmonaires élastiques, remplaçant l’intima normale. Ils occluent, complètement ou non, la lumière.
• Groupe 5 : ce groupe inclut des conditions hétérogènes avec des éléments anatomopathologiques différents dont l’étiologie est obscure ou multifactorielle.
Pathobiologie de l’hypertension pulmonaire
Divers éléments pathobiologiques caractérisent les divers groupes cliniques d’HTP.
• Groupe 1 : les processus qui initient les modifications pathobiologiques vues dans l’HTAP sont toujours imparfaitement connus, même si l’on sait que l’HTAP a une pathobiologie multifactorielle qui implique des voies biochimiques et des types cellulaires variés. L’augmentation des résistances vasculaires pulmonaires est due à divers mécanismes, incluant la vasoconstriction, un remodelage prolifératif et obstructif de la paroi vasculaire pulmonaire, de l’inflammation et de la thrombose.
• Groupe 2 : les mécanismes responsables de l’augmentation de la PAP sont multiples et incluent une transmission passive, en amont, de l’élévation pressionnelle (HTP passive postcapillaire, tableau 1) ; dans ces cas, le GTP et les résistances vasculaires pulmonaires sont dans les limites normales. Dans d’autres circonstances, l’élévation de la PAP est supérieure à celle de la P cap (GTP augmenté) et on observe aussi une augmentation des résistances vasculaires pulmonaires (HTP post-capillaire « réactive » ou « disproportionnée », tableau 1).
L’augmentation des résistances vasculaires pulmonaires est due à une augmentation du tonus vasomoteur des artères pulmonaires et/ou à un remodelage structural obstructif fixé des vaisseaux artériels pulmonaires résistifs.
• Groupe 3 : les mécanismes sont multiples et incluent une vasoconstriction hypoxique, un stress mécanique des poumons hyperinflatés, une perte de capillaires, de l’inflammation et les effets toxiques du tabagisme.
• Groupe 4 : le mécanisme pathobiologique le plus important est la non-disparition des masses emboliques aiguës qui ensuite entraînent de la fibrose conduisant à une obstruction mécanique des artères pulmonaires.
• Groupe 5 : la pathobiologie est obscure ou multifactorielle.
Génétique, épidémiologie et facteurs de risque de l’hypertension pulmonaire
• Groupe 1 : l’estimation la plus basse de la prévalence de l’HTAP et de l’HTAP idiopathique est de 15 et 6 cas/million de sujets adultes, respectivement ; l’estimation la plus basse de l’incidence annuelle de l’HTAP est de 2,4 cas/million de sujets adultes. Dans un registre français[ 3], 40 % des patients avec HTAP avaient une HTAP idiopathique, 4 % une HTAP héréditaire ; dans le sous-groupe des HTAP associées, 15 % avaient une connectivite (surtout une sclérodermie), 11 % une cardiopathie congénitale, 10 % une hypertension portale, 9 % avaient pris des anorexigènes et 6 % avaient une infection par le VIH. L’HTAP idiopathique est une maladie sporadique, sans aucune histoire familiale d’HTAP ni facteurs déclenchants ; lorsque l’HTAP survient dans un contexte familial, des mutations du gène BMPR2 sont détectées dans au moins 70 % des cas ; ces mutations peuvent aussi être détectées chez 11 à 40 % des cas apparemment sporadiques ; le gène BMPR2 encode le récepteur de type 2 de protéines de la morphogenèse osseuse ; parmi plusieurs fonctions biologiques, ces polypeptides sont impliqués dans le contrôle de la prolifération des cellules vasculaires ; des mutations d’autres récepteurs de ces substances, comme l’activin receptor-like kinase 1 et l’endogline, ont été identifiées, principalement chez les patients avec HTAP et des antécédents personnels ou familiaux de télangiectasies hémorragiques héréditaires (maladie de Rendu- Osler). De nombreux facteurs de risque de développement d’une HTAP ont été identifiés (HTAP associées : tableau 2 ; drogues et toxines : tableau 5).
• Groupe 2 : la prévalence de l’HTP chez les patients avec insuffisance cardiaque chronique augmente avec la progression de l’altération de la classe fonctionnelle ; il y a une HTP chez presque tous les patients avec atteinte mitrale très symptomatique et chez deux tiers des patients avec sténose aortique symptomatique.
• Groupe 3 : l’HTAP est très fréquente (> 50 %) dans la BPCO avancée ; la prévalence de l’HTP dans les maladies pulmonaires interstitielles est de l’ordre de 35 %.
• Groupe 4 : l’incidence de l’HTP après embolie pulmonaire est de 0,5-2 % ; on peut voir une HTP thromboembolique chronique chez des patients sans aucun épisode clinique d’embolie pulmonaire ou de thrombose veineuse profonde.
Hypertension artérielle pulmonaire (groupe 1)
C’est dans ce groupe que les avancées les plus importantes de la compréhension et du traitement ont été réalisées durant les dernières décennies. C’est aussi le groupe dans lequel l’HTP est le noeud des problèmes cliniques et peut être traitée par une thérapeutique médicamenteuse spécifique.
Diagnostic
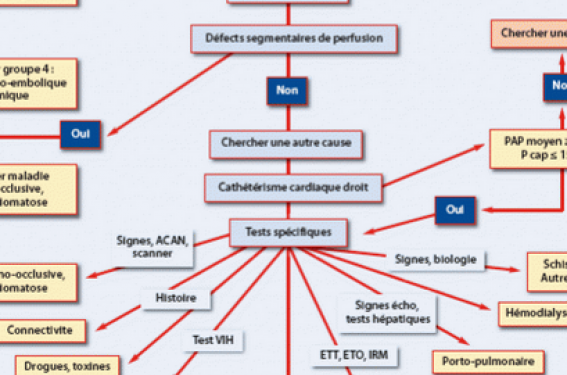
Les investigations face à une suspicion d’HTP ont pour but de confirmer le diagnostic, de clarifier le groupe clinique d’HTP et la cause spécifique, et d’évaluer l’importance de l’altération fonctionnelle et hémodynamique. La figure 1 présente l’algorithme diagnostique. Comme l’HTAP, et particulièrement l’HTAP idiopathique, est un diagnostic d’exclusion, cet algorithme peut être utilisé comme point de départ pour tous les cas de suspicion d’HTP.
Figure 1. Algorithme diagnostique.
Présentation clinique
Les symptômes de l’HTAP ne sont pas spécifiques ; ils incluent dyspnée, fatigue, angine de poitrine, syncope et distension abdominale. Les symptômes ne sont présents au repos que dans les cas très avancés. Les signes physiques d’HTAP incluent une augmentation de la composante pulmonaire de B2, un souffle systolique de régurgitation tricuspide, un souffle diastolique d’insuffisance pulmonaire et un B3 droit.
Une distension veineuse jugulaire, une hépatomégalie, des oedèmes périphériques, une ascite et des extrémités froides caractérisent les stades avancés. L’auscultation pulmonaire est habituellement normale. L’examen clinique peut aussi fournir des signes en faveur de la cause de l’HTP : télangiectasies, ulcération digitale et sclérodactylie dans la sclérodermie, crépitants inspiratoires dans la fibrose pulmonaire insterstitielle, angiomes stellaires, atrophie testiculaire et érythème palmaire en cas de maladie hépatique.
Électrocardiogramme
Il peut montrer une surcharge ou une hypertrophie ventriculaire droite, une dilatation atriale droite. Dans les stades avancés, il peut y avoir une arythmie supraventriculaire, notamment flutter atrial, mais aussi fibrillation atriale.
Radiographie thoracique
Chez 90 % des patients avec HTAP idiopathique, la radiographie thoracique est anormale : dilatation des artères pulmonaires, contrastant avec un élagage des vaisseaux périphériques, dilatation de l’oreillette droite et du ventricule droit.
Exploration fonctionnelle respiratoire et gazométrie artérielle
Dans l’HTAP, il y a habituellement une diminution de la capacité de diffusion du monoxyde de carbone et une réduction légère à modérée des volumes pulmonaires.
Échocardiographie
L’estimation de la PAP est basée sur la vitesse de régurgitation tricuspide.
D’autres anomalies échocardiographiques peuvent aider au diagnostic : la vitesse de régurgitation pulmonaire, un temps d’accélération de l’éjection du ventricule droit dans l’artère pulmonaire court. L’augmentation de la taille des cavités droites, une forme et un fonctionnement anormaux du septum interventriculaire, une augmentation de l’épaisseur pariétale du ventricule droit et une dilatation du tronc de l’artère pulmonaire suggèrent aussi une HTP, mais ces signes surviennent plus tard dans l’évolution de la maladie. Les propositions de critères arbitraires d’estimation de la présence d’une HTP selon la vitesse de régurgitation tricuspide, la PAP systolique et d’autres variables échographiques suggérant une HTP sont présentées dans le tableau 6.
L’échocardiographie peut être utile pour la détection de la cause de l’HTP (cardiopathie congénitale, etc.).
Scintigraphie pulmonaire de ventilation et de perfusion
L’examen doit être réalisé chez les patients avec HTP afin de dépister une HTP thromboembolique chronique, puisque sa sensibilité (90-100 %) est meilleure que celle de la tomodensitométrie. Dans l’HTAP, l’examen peut être normal, mais il peut aussi montrer de petits déficits périphériques de perfusion.
Tomodensitométrie à haute résolution, angiotomodensitométrie et angiographie pulmonaire
La tomodensitométrie à haute résolution est utile pour le diagnostic de maladie pulmonaire interstitielle, d’emphysème, de maladie veino-occlusive pulmonaire. L’angiotomodensitométrie des artères pulmonaires est utile pour déterminer s’il y a une HTP thomboembolique chronique accessible à la chirurgie. L’angiographie pulmonaire traditionnelle est cependant requise dans de nombreux centres s’occupant d’HTP thromboembolique chronique.
Imagerie par résonance magnétique cardiaque
Elle fournit une évaluation directe de la taille, la morphologie et le fonctionnement du ventricule droit et permet une évaluation non invasive du volume d’éjection, du débit cardiaque, de la distensibilité des artères pulmonaires et de la masse du ventricule droit.
Examens biologiques
Les examens biochimiques et hématologiques de routine et les tests de fonction thyroïdienne doivent être réalisés chez tous les patients. Les sérologies sont importantes pour détecter une connectivite, une infection par le HIV ou une hépatite.
Échographie abdominale
Elle est utile pour le diagnostic de cirrhose hépatique et/ou d’hypertension portale.
Cathétérisme des cavités cardiaques droites et tests de vasoréactivité
Le cathétérisme est requis pour confirmer le diagnostic d’HTAP, évaluer la sévérité de l’atteinte hémodynamique et tester la vasoréactivité de la circulation pulmonaire. Une P cap > 15 mmHg exclut le diagnostic d’HTAP précapillaire. Un des diagnostics différentiels les plus difficiles de l’HTAP est l’insuffisance cardiaque avec fraction d’éjection du ventricule gauche normale et dysfonction diastolique.
Les tests de vasoréactivité doivent être faits durant le cathétérisme afin d’identifier les patients qui peuvent bénéficier d’un traitement calcium-bloquant à long terme (tableau 7).
Les recommandations concernant le cathétérisme des cavités cardiaques droites et les tests de vasoréactivité sont résumées dans le tableau 8.
Algorithme diagnostique
Il est présenté dans la figure 1, et les éléments de prise en charge dans les tableaux 9 et 10.
Évaluation de la sévérité
Données cliniques, échocardiographiques et hémodynamiques
Malgré des variations inter-observateurs importantes, la classification fonctionnelle de l’OMS (tableau 11) est un prédicteur puissant de la survie. Chez des patients avec HTAP idiopathique ou héréditaire non traités, la durée médiane de survie est de 6 mois pour la classe IV, 2,5 ans pour la classe III, et 6 ans pour les classes I et II. Les âges extrêmes (< 14 ans ou > 65 ans), la mauvaise capacité d’exercice, les syncopes, les hémoptysies et l’insuffisance cardiaque droite sont eux aussi des éléments de mauvais pronostic dans l’HTAP idiopathique.
À l’échocardiographie, les meilleures variables pronostiques sont l’épanchement péricardique, l’augmentation de la pression atriale droite, l’index d’excentricité du ventricule gauche et l’index Doppler du ventricule droit. Au cathétérisme, sont pronostiques la saturation en oxygène dans l’artère pulmonaire, la pression atriale droite, le débit cardiaque, les résistances vasculaires pulmonaires et une réponse marquée au test de vasoréactivité.
Capacité d’exercice
Au test de marche de 6 minutes, une distance < 332 m ou < 250 m et une désaturation en oxygène > 10 % sont des éléments de mauvais pronostic.
À l’épreuve d’effort, dans l’HTAP idiopathique, en analyse multivariée, une consommation maximale d’oxygène < 10,4 ml/kg/min et une pression artérielle systolique pendant l’exercice < 120 mmHg sont des éléments de plus mauvais pronostic.
Marqueurs biochimiques
Plusieurs variables biochimiques apportent une information pronostique dans l’HTAP, mais leur utilité dans la pratique quotidienne n’est pas établie. Le dosage du BNP/NT-proBNP est recommandé pour la stratification initiale du risque et peut être envisagé pour la surveillance des effets du traitement.
Évaluation pronostique
Les paramètres sont listés dans le tableau 12, et le moment de leur réalisation dans le tableau 13.
Définition de l’état du patient
Il peut être :
– stable et satisfaisant : cela correspond aux paramètres de la colonne de gauche dans le tableau 12 ;
– stable et non satisfaisant : certains des paramètres de la colonne de gauche ne sont pas remplis ;
– instable et se détériorant : cela correspond aux paramètres de la colonne de droite.
Buts du traitement et stratégie de suivi
Ils sont résumés dans le tableau 14.
Traitements
Les recommandations concernant les mesures générales sont présentées dans le tableau 15, celles concernant les traitements « de soutien » dans le tableau 16, l’algorithme thérapeutique en ce qui concerne les médicaments spécifiques de l’HTAP dans la figure 2. Les classes de recommandation et les niveaux de preuve des diverses thérapeutiques sont présentés dans le tableau 17, et les définitions d’une réponse inadéquate dans le tableau 18.
Figure 2. Algorithme thérapeutique pour les patients avec hypertension artérielle pulmonaire (groupe 1 seulement).
Sous-groupes spécifiques de patients avec HTAP
Les tableaux 19 à 23 présentent les recommandations en ce qui concerne l’HTAP chez les enfants, l’HTAP associée à des shunts cardiaques congénitaux, l’HTAP associée à une connectivite, une hypertension portale ou une infection par le VIH.
Maladie veino-occlusive pulmonaire et hémangiomatose capillaire pulmonaire (groupe 1’)
Ce sont des maladies très rares, dont la prise en charge doit exclusivement être faite dans les centres spécialisées (tableau 24).
Hypertension pulmonaire due à une maladie du cœur gauche (groupe 2)
Les caractéristiques cliniques et échocardiographiques de l’HTAP associée à une dysfonction diastolique du ventricule gauche sont listées dans le tableau 25. Les recommandations pour la prise en charge de l’HTAP due à une maladie du coeur gauche sont résumées dans le tableau 26.
Hypertension pulmonaire due à une maladie pulmonaire et/ou à l’hypoxie (groupe 3)
Les symptômes et signes physiques de l’HTAP peuvent être difficiles à identifier chez des patients avec désordres respiratoires. De plus, dans la BPCO, les oedèmes périphériques peuvent ne pas être un signe d’insuffisance cardiaque droite mais être dus aux effets de l’hypoxémie et de l’hypercapnie sur le système rénine-angiotensinealdostérone. Enfin, une maladie du coeur gauche, assez souvent associée aux maladies pulmonaires, peut elle aussi contribuer à augmenter la PAP. Les recommandations sont résumées dans le tableau 27.
Hypertension pulmonaire thromboembolique chronique (groupe 4)
Une recherche de thromboembolie chronique doit être systématiquement faite en cas d’HTAP de cause inexpliquée. Une scintigraphie pulmonaire de ventilation et de perfusion doit être réalisée. Quand elle est normale, cela élimine une thromboembolie chronique. Les recommandations sont présentées dans le tableau 28.
Centre de référence pour l’hypertension pulmonaire
Les recommandations sont présentées dans le tableau 29.
Abréviations
BPCO : bronchopneumopathie chronique obstructive
CIA : communication interatriale
CIV : communication interventriculaire
GTP : gradient de pression transpulmonaire (PAP moyenne — P cap moyenne)
HTAP : hypertension artérielle pulmonaire
HTP : hypertension pulmonaire
PAP : pression artérielle pulmonaire
P cap : pression capillaire pulmonaire
Dans les tableaux présentant les recommandations, classe : classe de recommandation ; niveau : niveau de preuve
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
