


Publié le 11 oct 2011Lecture 7 min
Une dissection de l’aorte révélant un syndrome de Marfan
W. OUECHTATI, E. ALLOUCHE, H. BACCAR, R. KASRI, A. BELHENI, Hôpital Charles Nicolle, Tunis
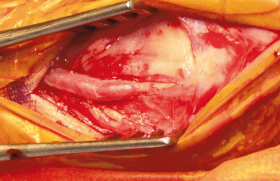
La dissection et la rupture aortiques sont les principales causes de morbi-mortalité dans le syndrome de Marfan. Ainsi, la chirurgie prophylactique de remplacement aortique est de plus en plus indiquée pour prévenir ces complications fatales. Nous rapportons le cas d’une patiente âgée de 36 ans, hospitalisée dans notre service pour prise en charge d’une dissection chronique de l’aorte thoraco-abdominale type B de Stanford sans signes de fissuration et d’un anévrisme de la crosse de l’aorte et devant l’aorte abdominale sous-rénale. Le diagnostic de syndrome de Marfan a été posé devant l’association d’une arachnodactylie, d’une scoliose rachidienne, la dissection aortique et le double anévrisme aortique. L’évolution s’est faite vers l’augmentation de taille des anévrismes compliqués de fissuration. L’indication opératoire a été alors posée, mais l’évolution a été rapidement fatale.
Le syndrome de Marfan est l’une des maladies génétiques les plus fréquentes ; selon les estimations, elle touche 1 sur 6 000 à 10 000 personnes dans le monde(1) ; en Europe, sa prévalence est de 1 pour 5 000. La transmission se fait selon le mode autosomique dominant à pénétrance complète, mais l’expression phénotypique est très variable entre les différentes familles atteintes et même au sein d’une seule famille(2). Le syndrome de Marfan associe généralement des manifestations oculaires, musculo-squelettiques et cardiovasculaires. L’atteinte cardiovasculaire détermine le pronostic, la cause essentielle du décès étant la survenue d’une dissection aortique, souvent précédée du développement d’un anévrisme(3,4).
Observation
Nous rapportons le cas d’une patiente âgée de 36 ans, opérée à l’âge de 14 ans pour genu varum, hospitalisée dans notre service pour la prise en charge d’une dissection chronique de l’aorte thoraco-abdominale type B de Stanford sans signes de fissuration.
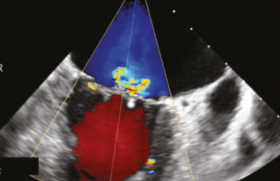
La dissection a été diagnostiquée 18 mois auparavant ; elle a bénéficié d’une IRM objectivant une dissection de l’aorte thoraco-abdominale allant de la crosse de l’aorte jusqu’au niveau de la naissance des artères rénales et épargnant l’aorte thoracique ascendante qui reste de calibre normal (2,5 cm) (figure 1), un anévrisme fusiforme de la crosse de l’aorte de 4,6 cm et un anévrisme de l’aorte abdominale sous-rénale de 4,4 cm.
Figure 1. Dissection de l’aorte thoraco-abdominale type B.
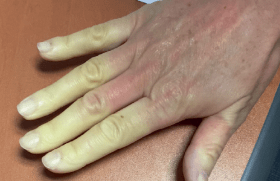
Par ailleurs, la patiente présentait une hyperlaxité musculo-tendineuse (figure 2) et une scoliose rachidienne.
Ces données cliniques et radiologiques ont permis de porter le diagnostic de syndrome de Marfan compliqué d’une dissection aortique.
Figure 2. Arachnodactylie.
Devant la stabilité hémodynamique, l’absence de signes de fissuration et la possibilité de contrôler la pression artérielle, on a opté, après discussion médicochirurgicale, pour le traitement médical (repos strict, surveillance et traitement antihypertenseur par bêtabloquant) et d’opérer ultérieurement.
À l’examen : l’état hémodynamique était stable, la tension artérielle était symétrique aux 2 bras et il n’y avait pas de souffle audible à l’auscultation cardiaque.
Durant son hospitalisation, la patiente a présenté des douleurs lombaires gauches et du flanc gauche, sans signes de choc, motivant la pratique d’un angio-scanner qui a mis en évidence une augmentation de la taille des anévrismes : à l’étage thoracique 8 cm et à l’étage abdominal 11 cm, présence d’un épaississement pariétal en regard du vrai chenal, spontanément hyperdense, évoquant un hématome pariétal et un épanchement abdominal de faible abondance en regard du flanc gauche (figures 3 à 5). Devant ces signes de fissuration, il a été décidé de la transférer en chirurgie cardiovasculaire pour l’opérer en urgence. En préparant son transfert, la patiente a décrit une sensation de chaleur interne suivie immédiatement d’un arrêt cardiorespiratoire non récupéré, et la patiente est décédée d’une rupture aortique très probable.
Figure 3. Hématome pariétal.
Figure 4. Hématome pariétal.
Figure 5. Augmentation de la taille de l’anévrysme abdominal.
Discussion
Le syndrome de Marfan est une maladie héréditaire du tissu conjonctif de transmission autosomique dominante. Il résulte d’une mutation du gène FBN1 situé sur le chromosome 15 et codant pour la fibrilline 1, protéine essentielle du tissu conjonctif, ce qui cause la dégénérescence et l’affaiblissement progressif du tissu conjonctif(3) ; 85 % des cas de Marfan sont causés par l’une de plus de 600 mutations connues du FBN1. La physiopathogénie de cette maladie consiste en une diminution de la résistance des différents tissus et organes à la pression et à la torsion car la fibrilline est une protéine importante dans l’ultrastructure des fibres d’élastine(2).
Le syndrome de Marfan associe généralement des atteintes ophtalmologiques à type de luxation du cristallin, des atteintes squelettiques, telles une dolichosténomélie (un excès de croissance des membres), une arachnodactylie (doigts longs et fins), des déformations rachidiennes assez fréquentes (scoliose, cyphose) et une hyperlaxité ligamentaire, des atteintes pulmonaires (emphysème) et cardiovasculaires. Ces dernières sont généralement faites de dilatation de la racine de l’aorte, d’une insuffisance aortique, prolapsus mitral et de dissection de l’aorte(4).
Notre patiente présentait une dolichosténomélie, une arachnodactylie, une hyperlaxité ligamentaire, une scoliose, un double anévrisme de l’aorte et la dissection aortique.
L’atteinte cardiovasculaire, cliniquement présente dans 60 à 90 % des cas, conditionne la sévérité du syndrome de Marfan, puisque les complications peuvent mettre la vie en péril. En effet, 34 % des Marfan présenteront de sérieuses complications cardiovasculaires nécessitant le recours à la chirurgie dans les dix premières années suivant le diagnostic(5).
Ainsi, plus de 90 % des décès dont l’étiologie est identifiée impliquent le système cardiovasculaire, les causes essentielles du décès étant la survenue d’une dissection compliquant un anévrisme de la racine aortique ou la rupture de l’aorte fragilisée(6).
Le prolapsus mitral est la valvulopathie la plus fréquente chez les Marfan, elle est présente dans 35 % à 100 % des cas(7). Notre patiente ne présentait pas cette anomalie.
Environ 70 % des patients porteurs d’un syndrome de Marfan présentent une dilatation de la racine aortique, le plus souvent modérée. Cette proportion augmente avec l’âge et justifie une intervention chirurgicale chez 25 % de ces patients. En l’absence de traitement, 90 % des patients décèdent de complications cardiovasculaires, dont 80 % de rupture aortique. L’association d’un traitement par bêtabloquants et de la chirurgie prophylactique à partir d’un diamètre de la racine aortique de 50 mm, a permis de prolonger de 30 ans la survie des patients porteurs d’un syndrome de Marfan chez qui l’espérance de vie ne dépassait pas 32 ans comparativement à 70 ans actuellement(1). Une intervention prophylactique précoce offre une survie de 97 % à 10 ans (versus 31 % à 10 ans après chirurgie en urgence)(8).
Dans notre observation, le calibre de l’aorte ascendante était normal (2,5 cm) et le diagnostic de syndrome de Marfan n’a été évoqué qu’après la survenue de la dissection.
Dix pour cent des patients porteurs d’un syndrome de Marfan sont opérés en urgence d’une dissection de l’aorte ascendante (Marfan non diagnostiqué et/ou non suivi). Ces patients représentent 50 % des dissections aortiques aiguës chez les moins de 40 ans(1).
La dissection de l’aorte descendante est une complication plus rare. Dans 80 % des cas, elle correspond à l’extension d’une dissection de l’aorte ascendante.
Dans le cadre de notre observation, la patiente présentait une dissection de l’aorte thoraco-abdominale allant de la crosse de l’aorte jusqu’au niveau de la naissance des artères rénales, épargnant l’aorte thoracique ascendante.
Moins de 20 % des dissections aortiques survenant chez les Marfan sont de type B, comme chez notre patiente. Les dissections aortiques aiguës de type B doivent être traitées médicalement, sauf en cas d’ischémie (mésentérique ou membres inférieurs) ou de rupture(8).
Pour l’aorte thoraco-abdominale, l’indication chirurgicale élective repose essentiellement sur l’évolution anévrismale d’une dissection chronique : l’indication est formelle au-delà de 65 mm de diamètre ; elle peut être également discutée à partir d’un anévrisme de 50 mm de diamètre dont la croissance est supérieure à 5 mm/an (figure 6).
Figure 6. Arbre décisionnel : conduite à tenir devant un syndrome de Marfan avec atteinte aortique(1).
Pour notre patiente, le diamètre de l’aorte anévrismale disséquée était de 4,6 cm au moment du diagnostic, et s’est agrandi à 5,9 cm au bout de 18 mois, soit une augmentation > 6 mm/an, d’où l’indication opératoire.
Le diamètre aortique constitue donc une indication opératoire, mais son augmentation ou celle de la taille du faux chenal sont aussi des paramètres qui poussent à opérer le patient, même asymptomatique. En postopératoire, le pronostic peut être nettement amélioré et le risque opératoire reste toujours inférieur au risque spontané de rupture(9). Certaines équipes rapportent une mortalité de 2 % à 1 mois(10). En dehors des grands centres, la mortalité de cette chirurgie est de 10 à 42 %. En postopératoire, la paraplégie et l’insuffisance rénale constituent les complications les plus fréquentes, chez 4 à 32 % et 4 à 37 % des patients, respectivement(9).
En pratique
La dissection et la rupture aortiques sont les principales causes de morbi-mortalité dans le syndrome de Marfan. La chirurgie prophylactique de remplacement aortique est de plus en plus indiquée pour prévenir ces complications fatales.
Le rôle du traitement percutané par stent des dissections de type B chez les porteurs d’un syndrome de Marfan n’est pas encore bien établi. Cette procédure n’est qu’une alternative pour réduire le nombre de chirurgies aortiques chez les Marfan qui présente un taux de mortalité de 31 %(1).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
