


Rythmologie et rythmo interventionnelle
Publié le 02 sep 2008Lecture 9 min
Le calcium intracellulaire : un élément-clé de la physiopathologie de la mort subite
J.-L. PASQUIÉ, S. RICHARD, CHU Montpellier

Le Printemps de la cardiologie
Les arythmies ventriculaires responsables de mort subite cardiaque (MSC) surviennent essentiellement à un stade précoce de l’insuffisance cardiaque et sont de ce fait souvent qualifiées « d’imméritées ». Au cours de la dernière décennie, il est devenu de plus en plus évident que des altérations des mouvements calciques intracellulaires peuvent être la source d’arythmies ventriculaires par le biais d’une modulation de l’activité de canaux ioniques régulés par le calcium (Ca2+).
Le canal ryanodine sensible de type 2 (RyR2) est une protéine du réticulum sarcoplasmique (RS) des cardiomyocytes qui est essentielle à la régulation des mouvements calciques intracytoplasmiques. Son dysfonctionnement isolé est maintenant reconnu comme étant la cause de tachycardies ventriculaires polymorphes catécholergiques (TVPC). Il s’agit d’une anomalie de fonctionnement qui apparaît également progressivement au cours du développement de l’insuffisance cardiaque, quelle que soit son origine, et qui peut être alors à l’origine d’arythmies ventriculaires par surcharge calcique intracellulaire en diastole.
Mort subite cardiaque et surcharge calcique : le lien
Au cours de chaque battement cardiaque, la dépolarisation de la membrane cellulaire des cardiomyocytes ouvre les canaux calciques de type L situés au niveau des tubules transverses (tt), ce qui produit un influx de Ca2+ extracellulaire dans le cytoplasme. Ce « courant » calcique déclenche une libération massive de Ca2+ stocké dans le RS en provoquant une ouverture synchronisée de protéines appelées RyR2 (Ca2+-induced Ca2+-release). Le RyR2 est un complexe macromoléculaire très régulé, dont le pore est un homotétramère composé de quatre sous-unités associées. Son ouverture et sa fermeture sont régies par le Ca2+. Une augmentation du Ca2+ cytosolique (< 0,1 µM en diastole) à 1 µM ouvre le RyR2. Des concentrations beaucoup plus élevées l’inactivent (le ferment), ce qui arrive suite à la libération massive de Ca2+ du RS en systole. En plus de nombreux mécanismes endogènes de régulation par différentes protéines auxiliaires et mécanismes de phosphorylation, le RyR2 est régulé par de multiples agents pharmacologiques comprenant, entre autres, la ryanodine (par définition), la caféine, les aminophyllines, le dantrolène, le rouge de ruthénium, des métaux lourds, des anesthésiques…
Le mécanisme de « Ca2+-induced Ca2+-release » amplifie considérablement le signal initial de Ca2+ entrant via les canaux calciques de type L et fournit la quantité de Ca2+ nécessaire pour l’activation des protéines contractiles pendant la systole. Cette séquence d’événements conditionne le processus de « couplage excitation-contraction », mécanisme fondamental pour le développement de la contraction cardiaque. La relaxation se produit lorsque le Ca2+ cytoplasmique est restocké dans le RS par la pompe Ca2+-ATPase SERCA et/ou expulsé de la cellule par l’échangeur Na+-Ca2+ (NCX) qui présente la particularité d’être électrogénique. Trois ions Na+ entrent dans la cellule pour permettre l’extrusion d’un ion Ca2+, la charge nette entrante générant une dépolarisation spontanée en diastole (figure 1).
Si au niveau de l’organe, on considère que les arythmies ventriculaires (figure 2) dans l’insuffisance cardiaque sont essentiellement dues à des phénomènes de réentrées, au niveau cellulaire, elles peuvent provenir d’un déséquilibre entre courants dépolarisants et courants repolarisants de la phase de plateau du potentiel d’action (PA). L’allongement du PA résulte en grande partie d’une diminution de courants potassiques repolarisants et, à un moindre degré, d’un ralentissement de l’inactivation du courant calcique (qui dépend de la charge calcique du RS) pendant la systole. L’insuffisance cardiaque est également associée à des changements importants des mouvements intracellulaires de Ca2+. Ces changements incluent une diminution du contenu en Ca2+ du RS, à cause :
• d’une réduction du restockage du Ca2+ dans le RS, liée à une diminution de l’expression de la SERCA ;
• d’une fuite du Ca2+ hors du RS, occasionnée par des ouvertures inopportunes des RyR2 en diastole.
Figure 1. (A) Au cours de la systole, le potentiel d’action (PA) se propage le long de la membrane plasmique (phase 1). La dépolarisation ouvre les canaux calciques de type L (sensibles aux « antagonistes calciques »), localisés dans des invaginations de la membrane appelées tubules transverses (tt) : du Ca2+ du milieu extracellulaire entre dans la cellule (phase 2). Ce Ca2+ induit l’ouverture des RyR2, qui libèrent alors une quantité importante du Ca2+ contenu dans le réticulum sarcoplasmique (RS) générant une augmentation transitoire du Ca2+ dans la cellule (phase 3). Ce Ca2+ active alors la machinerie contractile et la contraction (phase 4) ; (B) au cours de la phase de relaxation, le Ca2+ relaché par les protéines contractiles (phase 5) est restocké dans le RS via la SERCA (phase 6) et/ou rejeté dans le milieu extracellulaire via le NCX (phase 7) pour un retour à l’état diastolique.
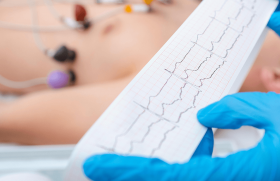
Figure 2. Tracé ECG chez un patient de 70 ans porteur d’une cardiopathie dilatée d’origine valvulaire (prothèse de St Jude aortique pour insuffisance aortique) avec FEVG à 25 %. Un DAI a été implanté 6 ans auparavant pour une mort subite récupérée. En l’absence de troubles métaboliques ou de surdosage médicamenteux, des ESV apparaissent isolées ou en doublets (tracé A) puis, quelques dizaines de minutes plus tard (tracé B), une deuxième morphologie d’ESV d’axe opposé apparait puis induit un flutter ventriculaire nécessitant un choc de cardioversion interne par le DAI (CV).
La diminution du Ca2+ du RS participe à la diminution du transitoire calcique et de la contraction associée. La diminution de la reprise du Ca2+ cytoplasmique par la SERCA rend compte de la phase de relaxation ralentie de la contraction, malgré une augmentation d’expression et/ou de fonction du NCX qui a pour objectif d’extruder une partie du Ca2+ intracellulaire et, donc, de limiter toute surcharge calcique délétère.
Le RyR2 joue un rôle clé dans le contrôle de l’homéostasie calcique intracellulaire puisque sa fermeture, normale pendant la diastole, empêche la sortie du Ca2+ du RS et favorise, de ce fait, la charge pour la systole qui suit. Par conséquent, une incontinence chronique du RyR2 occasionne une fuite chronique de Ca2+ avec un impact majeur, non seulement sur la contraction mais aussi sur le déclenchement des arythmies induites par toute surcharge calcique intracellulaire via les post-dépolarisations tardives (DADs).
TVPC : un modèle presque « expérimental » de fuite du RyR2
Les tachycardies ventriculaires polymorphes catécholergiques (TVPC) sont une cause rare de syncope et de mort subite cardiaque (MSC) chez des enfants, des adolescents et de jeunes adultes présentant un cœur pourtant structurellement normal. Cette maladie génétique rare, souvent familiale, est caractérisée par des arythmies ventriculaires déclenchées par l’exercice physique ou le stress émotionnel. Le premier événement syncopal se produit généralement dans l’enfance avec un âge moyen de 8 ans, c’est-à-dire plus jeune que l’âge moyen pour la première syncope chez les patients avec un QT long congénital (QTLC). Si les patients avec un QTLC développent des torsades de pointe (TdP), les patients « TVPC » présentent une tachycardie ventriculaire bi-directionnelle, caractérisée par une rotation de 180° battement à battement de l’axe électrique du QRS.
Contrairement au QTLC, le diagnostic de TVPC ne peut être basé sur l’ECG de repos car aucune anomalie n’est présente. Les TVPC sont généralement associées à un risque élevé de MSC ; 80 % des patients développent différents symptômes, incluant fibrillation ventriculaire, tachycardie ventriculaire ou syncope, avant 40 ans, avec une mortalité globale rapportée de 40 %. Les investigations génétiques ont démontré que les TVPC sont associés à des mutations de gènes codant pour des protéines impliquées dans la régulation des mouvements calciques intracellulaires. Deux variantes de la maladie ont été identifiées : une forme dominante autosomale liée à des mutations du gène codant pour le récepteur à la ryanodine (RyR2) cardiaque, et une forme récessive associée à des mutations du gène codant l’isoforme cardiaque de la calséquestrine (CASQ2), une protéine fixant le Ca2+ avec une faible affinité, associée au RyR2 sur le côté luminal du RS. RyR2 et CASQ2 sont deux protéines très impliquées dans la régulation du Ca2+ intracellulaire. De nombreuses mutations du RyR2 ont été identifiées à ce jour.
Bien que le RyR2 n’ait aucun rôle électrogénique direct, il influe sur la signalisation électrique et produit des troubles du rythme via le Ca2+ qu’il libère de manière inopportune pendant la diastole.
Bases mécanistiques des ouvertures aberrantes du RyR2
Comment l’hyperactivité des RyR2 pendant la diastole peut-elle être traduite en signalisation aberrante et en induction d’activité électrique anormale de type DADs ? Une interprétation commune est que, à la fois dans les TVPC et dans l’insuffisance cardiaque, le substrat principal des DADs est la surcharge calcique intracellulaire – liée en particulier à une ouverture anormale des RyR2 en diastole – qui active un courant entrant transitoire (Iti) via le NCX électrogénique (figure 3). La surcharge calcique intracellulaire pourrait également inhiber le courant IK1, dans l’insuffisance cardiaque – et peut-être aussi dans les TVPC. IK1 a un rôle fondamental dans le contrôle du potentiel de membrane diastolique des cardiomyocytes et permet la bonne repolarisation du PA au cours de la phase tardive du plateau. On pense que l’activation concomitante d’Iti et la diminution d’IK1 par la surcharge calcique agissent de manière synergique pour déstabiliser le potentiel diastolique de repos de la membrane cellulaire, réunissant ainsi des conditions favorables pour des dépolarisations spontanées et inopportunes, et ainsi l’occurrence de DADs.
Figure 3. (A) Une incontinence du RS, induite par des ouvertures spontanées et inopportunes du RyR2 au cours de la diastole, produit une augmentation du Ca2+ libre intracellulaire. Ce Ca2+ est extrudé par le NCX qui génère un courant sodique entrant dépolarisant. L’intensité de la fuite calcique via les RyR2 détermine l’amplitude de la dépolarisation membranaire et peut déclencher des vagues calciques et des PA spontanés qui sont sources d’arythmie. (B) Toute situation qui augmente la charge calcique du RS (via une stimulation b-adrénergique, par exemple) ou qui déstabilise l’état fermé des RyR2 (abaissement de la sensiblité au Ca2+ pour l’ouverture en diastole dans le cas des TVPC ; hyperphosphorylation liée à une stimulation b-adrénergique chronique dans l’insuffisance cardiaque) induit – ou révèle des risques latents – d’arythmies ventriculaires Ca2+-dépendantes.
Une stimulation sympathique (b-adrénergique) préservée (ou éventuellement une activation parallèle via d’autres récepteurs impliquant la voie de phosphorylation par la protéine kinase AMPc-dépendante) est impliquée dans le déclenchement des arythmies Ca2+-dépendantes des TVPC et de l’insuffisance cardiaque. On s’attend, en effet, à ce que la stimulation b-adrénergique (ou toute stimulation équivalente) augmente la charge en Ca2+ du RS avec, comme conséquence directe, une augmentation de la fuite du Ca2+ dans le cytoplasme via les RyR2 perméables en diastole. Ainsi, un risque latent de déclenchement de DADs peut donc devenir avéré via la stimulation‚ b-adrenergique ou la voie PKA-dépendante. Une augmentation de la sensibilité du RyR2 au Ca2+ de la lumière du RS (pour les TVPC) et/ou de la charge du RS en amont constituent des mécanismes probables. Par ailleurs, bien que cela soit controversé, un processus d’hyperphosphorylation du RyR2 par la PKA pourrait aussi être impliqué pour expliquer l’instabilité du canal en diastole.
Perspectives cliniques
Dans l’insuffisance cardiaque, plusieurs traitements pharmacologiques pourraient avoir un effet bénéfique sur le dysfonctionnement du RyR2 :
• par exemple, les bêtabloqueurs et le valsartan (bloqueur des récepteurs à l’angiotensine II) amélioreraient la fonction de libération calcique de RyR2, peut-être en normalisant sa phosphorylation, ou encore la stœchiométrie du complexe macromoléculaire et la prévention de la fuite calcique. Les bêtabloqueurs sont aussi utilisés pour traiter les TVPC ;
• à côté de ces approches pharmacologiques indirectes, des médicaments potentiels comme le JTV519 (benzothiazépine 1,4) et ses dérivés se fixent sur le RyR2, normalisent son activité et réduisent la fuite anormale de Ca2+, améliorant ainsi la fonction contractile et empêchant les arythmies ventriculaires ;
• l’anti-oxydant édavarone pourrait également améliorer la stabilité du canal par un mécanisme complexe de correction d’interactions défectueuses au cœur du RyR2.
En pratique
La découverte et le développement de nouveaux médicaments anti-arythmiques, capables d’empêcher des ouvertures inopportunes du RyR2 en diastole et, donc, les arythmies qui dépendent de la surcharge calcique intracellulaire, représentent actuellement un défi important.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
