


Publié le 14 sep 2010Lecture 16 min
Spécificités des cardiopathies congénitales de l'adulte
P. AMEDRO, CHU de Montpellier
Le Printemps de la cardiologie
Toutes les études épidémiologiques ont constaté sur les deux dernières décennies une augmentation significative de la prévalence des cardiopathies congénitales à l’âge adulte (GUCH). Dans la majorité des pays développés leur prévalence égale désormais celle de la population pédiatrique. L’étude canadienne de Marelli, publiée en 2007 (tableau 1), montre que la prévalence des cardiopathies congénitales chez l’adulte a augmenté de 85 % entre 1985 et 2000, passant de 0,21 à 0,38/1 000 habitants, alors qu’elle ne passait que de 1,19 à 1,45 chez les sujets de moins de 18 ans.
Chez les enfants (< 18 ans) comme chez les adultes, la prévalence des cardiopathies congénitales (CC) est en augmentation, même si l’incidence à la naissance reste globalement stable, autour de 0,8 %. Plusieurs explications sont évoquées : performance du diagnostic prénatal (la France a été le premier pays à montrer l’impact du diagnostic prénatal sur l’incidence de l’hypoplasie du coeur gauche), dépistage échocardiographique précoce des CC mineures, progrès chirurgicaux (CEC chez le nouveau-né, programmes palliatifs de type coeur univentriculaire), âge maternel plus avancé, etc.
En France, la cardiologie congénitale de l’adulte concernerait environ 200 000 patients. Dans la majorité des régions, une collaboration plus ou moins structurée est assurée entre cardiopédiatres, cardiologues, rythmologues, cathétériseurs, radiologues et chirurgiens cardiaques. Les véritables unités spécialisées sont toutefois très rares, contrairement aux pays anglosaxons qui ont anticipé le flux des GUCH. Ainsi, la transition vers l’âge de 18 ans entre la cardiopédiatrie et la cardiologie adulte est une étape cruciale où le malade ne doit pas être « perdu de vue ». Les meilleurs centres américains constatent cette fuite des malades une fois la période pédiatrique passée, pour des motifs variés.
Le « catalogue » des cardiopathies congénitales est souvent indigeste pour le cardiologue « généraliste », car il représente pour lui une association de maladies rares éloignées de sa pratique quotidienne et peu évoquées lors de sa formation initiale. Pour diminuer cet obstacle on peut choisir de diviser les cardiopathies congénitales en :
– CC simples, diagnostiquées et prises en charge de façon relativement proche chez l’enfant et l’adulte,
– CC complexes qui nécessitent impérativement une prise en charge multidisciplinaire spécialisée.
Nous traiterons ici trois exemples de CC en apparence simples : les CIA vieillies, les CIV vieillies et la tétralogie de Fallot opérée. Ces trois cardiopathies congénitales représentent à elles seules la majorité des problèmes rencontrés par un cardiologue « généraliste ».
Les cardiopathies congénitales complexes ne seront pas traitées ici ; elles sont habituellement prises en charge, conformément aux objectifs du plan maladies rares, par les équipes pluridisciplinaires des centres de référence et de compétence, récemment labellisés en 2008.
Les communications interauriculaires (CIA) « vieillies»
La CIA de type foramen ovale perméable (FOP) peut être révélée par un accident vasculaire cérébral, des migraines, ou découverte fortuitement sur une échographie ; un FOP n’entraine pas d’hyperdébit pulmonaire, donc ne s’accompagne pas de souffle organique. On tente actuellement de mieux préciser les indications de fermeture par cathétérisme interventionnel.
La CIA ostium secundum, ou de la fosse ovale, est centrale, respecte le septum primum et est de loin la plus fréquente, avec une prédominance féminine. Elle est rarement symptomatique chez l’enfant, s’accompagne typiquement d’un souffle systolique au foyer pulmonaire avec dédoublement du B2 ; chez l’adulte, le souffle s’atténue, et cliniquement tout est possible, en fonction du retentissement de cette CIA « vieillie » (insuffisance cardiaque, trouble du rythme auriculaire). La CIA secundum se ferme le plus souvent par cathétérisme interventionnel via une prothèse larguée sous contrôle ETO. Les indications de fermeture percutanée d’une CIA sont :
- CIA ostium secundum sans extension vers la veine cave inférieure ;
- diamètre étiré au ballonnet gonflable (« sizing ballon ») > 40 mm ;
- rebord circonférentiel d’au moins 4 mm, sauf en avant et en haut, sous l’aorte ;
- absence de proximité des valves auriculo-ventriculaires.
En pratique, en cas de CIA très large ou sans rebord postérieur, la fermeture est effectuée par le chirurgien, souvent par thoracotomie postérieure.
La CIA sinus venosus est très basse, près de l’abouchement de la veine cave inférieure (VCI), avec parfois un retour veineux pulmonaire anormal partiel de la veine pulmonaire supérieure droite dans l’abouchement de la VCI à l’oreillette droite. La cure est chirurgicale. Le TDM précise le retour veineux pulmonaire.
La CIA ostium primum, très haute, au-dessus des valves auriculo-ventriculaires, fait partie des canaux atrio-ventriculaires et est en général diagnostiquée chez l’enfant. La cure est chirurgicale.
La CIA de type « sinus coronaire » est exceptionnelle et constitue un piège diagnostique : il s’agit d’une déhiscence du toit du sinus coronaire qui communique avec l’oreillette gauche (OG) ; l’échographie de contraste est une aide au diagnostic.
Chez l’adulte, en pratique, la CIA « vieillie » pose deux problèmes :
- anatomique : la CIA est plus difficile à diagnostiquer que chez l’enfant ; cliniquement, le souffle au foyer pulmonaire diminue et échographiquement la voie sous-costale est difficile, alors que chez l’enfant elle permet une visibilité parfaite de tout le septum interauriculaire, des veines caves et des veines pulmonaires supérieures.
Deux signes indirects sont à rechercher impérativement :
un ventricule droit (VD) non dilaté et un mouvement normal du septum interventriculaire permettent d’éliminer quasiment toujours une CIA à shunt significatif. L’ETO est donc plus souvent réalisée chez l’adulte alors que, chez l’enfant, elle n’est effectuée que lors d’une fermeture par cathétérisme interventionnel.
L’analyse des pressions pulmonaires est impérative, ainsi que la fonction VD.
- hémodynamique : l’indication de fermeture d’une CIA « vieillie » est plus compliquée et doit être réfléchie. Schématiquement, on distingue 3 situations (tableau 2) qui correspondent à l’histoire naturelle de l’HTAP dans les shunts gauchedroite.
– Dans le groupe 1, le shunt G- >D est significatif : l’échocardiographie est l’examen de référence pour évaluer ce shunt : VD dilaté (VDd/VGd = 1/2 à 1), Qp/Qs > 2, PAP élévées par hyperdébit pulmonaire (loi de Poiseuil : PAPm = Qp x RVP + POG) ; le plus souvent, les PAP sont de l’ordre de 1/3 à 1/2 des pressions systémiques soit, en pratique, des PVD par le flux d’insuffisance tricuspide entre 40 et 60 mmHg. Ici, l’indication de fermeture de la CIA est consensuelle, d’autant plus si elle est symptomatique.
– Dans le groupe 3, ou syndrome d’Eisenmenger, l’HTAP est fixée.
Après plusieurs décennies d’hyperdébit pulmonaire par le shunt G->D, le « shear stress » a entrainé des lésions endothéliales sur l’arbre artériel pulmonaire.
Comme pour l’HTAP idiopathique, apparaissent une prolifération des cellules musculaires lisses, une augmentation de la matrice extracellulaire et une thrombose intravasculaire ; ces phénomènes perturbent l’équilibre vasodilatation/vasoconstriction pulmonaire et prolifération/antiprolifération (figure 1) et aboutissent à l’augmentation des RVP, de façon autonome par rapport au shunt. À un stade avancé, ce shunt s’inverse, le patient devient alors cyanosé.
Figure 1. Physiopathologie de l’HTAP dans les shunts G->D.
La CIA fonctionne comme une soupape de sécurité lorsque RVP > RVS pour assurer un débit systémique. Sans CIA, comme pour l’HTAP primitive, cette situation hémodynamique n’est pas viable.
Ainsi les patients « Eisenmenger » ont une médiane de survie supérieure à celle des autres causes d’HTAP. La physiopathologie de l’HTAP issue des shunts G->D rejoignant celle de l’HTAP primitive (Celermajer et al. Circulation 1994 ; Rabinovitch et al. Lab Invest 1986), ces malades peuvent bénéficier des mêmes thérapeutiques (bosentan, sildénafil), mais le bénéfice à long terme reste à démontrer.
– Le groupe 2 constitue une « zone grise », entre ces deux situations extrêmes et caricaturales des groupes 1 et 3. Plusieurs questions se posent : HTAP de débit ? HTAP réversible ? RVP trop élevées ? CIA à fermer ? Ici le cathétérisme cathétérisme cardiaque est indispensable : mesure des pressions et résistances vasculaires pulmonaires, calcul du shunt (par le principe de Fick, la mesure du débit par thermodilution n’étant pas fiable dans les shunts), test de vasoréactivité (NO, O2). Si les RVP indexées sont < 6 UWood/m2 et que le rapport des résistances RVP/RVS est < 0,35, la CIA peut être fermée. La réversibilité au test de vasoréactivité est définie par une baisse des PAPm d’au moins 10 mmHg avec des PAPm qui deviennent < 40 mmHg et un débit qui est conservé ou augmenté. En cas de doute, il convient de ne pas fermer la CIA. C’est dans ces situations exceptionnelles que peuvent être effectuées des biopsies pulmonaires : l’étude histologique permet, dans des centres d’anatomopathologie spécialisés, de grader (classification de Heath-Edwards) le niveau de l’HTAP en associant plusieurs critères (hyperplasie musculaire, prolifération intimale, dilatation, nécrose, etc.) ; au-delà du grade III, l’évolution vers un syndrome d’Eisenmenger est presque inéluctable et la fermeture du shunt est contre-indiquée.
Certains évoquent la possibilité de traiter ces patients de la « zone grise » par les nouvelles molécules agissant sur le remodelage vasculaire pulmonaire (type bosentan) afin de passer du groupe 2 au groupe 1, mais cette vision séduisante est peut être utopique.
Les communications interventriculaires « vieillies » (CIV)
Il est très rare qu’une CIV soit révélée à l’âge adulte car le souffle est souvent intense, typiquement systolique et irradiant en rayon de roue. Là encore, le cardiologue devra préciser l’anatomie et l’hémodynamique de cette CIV.
La classification anatomique de 1980 proposée par RH Anderson décrit les CIV en observant le septum interventriculaire par le ventricule droit.
Les CIV musculaires sont complètement entourées de septum interventriculaire musculaire ; à la naissance, elles représentent les deux tiers des CIV. Elles sont souvent trabéculées, centrales ou apicales et de petite taille. Lorsqu’elles sont multiples, apicales, on parle de « fromage de gruyère ». Elles se ferment souvent spontanément.
Les CIV périmembraneuses représentent un tiers des CIV à la naissance ; elles sont bordées par du tissu fibreux situé à la jonction des orifices valvulaires et du corps central fibreux. Elles peuvent se fermer spontanément (complètement ou partiellement) par un anévrysme du septum membraneux, à partir du tissu tricuspide. Elles peuvent s’étendre vers le septum d’admission, le septum musculaire et le septum infundibulaire. En coupe parasternale petit axe passant par les sigmoïdes aortiques, le Doppler couleur donne la direction du shunt (flux rouge) entre 19 h et 12 h si on prend l’anneau aortique comme « horloge ».
Les CIV infundibulaires sont sous-pulmonaires et plus fréquentes en Asie. En coupe parasternale petit axe passant par les sigmoïdes aortiques, le Doppler couleur donne la direction du shunt (flux rouge) entre 12 h et 15 h. Elles sont parfois associées à une fuite aortique, (syndrome de Pezzi-Laubry), ce qui constitue alors une indication chirurgicale. En général, elles ne se ferment pas spontanément.
Les CIV de la chambre d’admission, postérieures, peuvent entrer dans l’entité « canal atrio-ventriculaire » ; elle sont en général larges, ne se ferment pas spontanément et les lésions associées doivent être recherchées.
Les CIV cono-ventriculaires sont sous-aortiques, comportent une déviation du septum conal et un malalignement aortique (CIV de la tétralogie de Fallot) ; elles ne se ferment pas spontanément.
L’évaluation hémodynamique des CIV à tout âge, et a fortiori à l’âge adulte, est capitale.
Rares sont les CIV d’indication chirurgicale chez l’adulte car les décisions ont été prises lors de la période pédiatrique.
Quatre classes hémodynamiques sont classiquement décrites :
• Classe I : CIV restrictives, de petite taille (< 5 mm en général).
C’est la maladie de Roger. Le patient a un gros souffle et est parfaitement asymptomatique. Le gradient trans-septal (Doppler continu sur la CIV, dans l’axe du shunt) est élevé (> 5 m/s), il n’y a pas de signe direct ou indirect d’hyperdébit pulmonaire (Qp/Qs = 1, VG non dilaté). En général l’évaluation échographique suffit, il n’y a pas d’indication thérapeutique ; seules les CIV infundibulaires avec apparition d’une fuite aortique (syndrome de Pezzi-Laubry) peuvent justifier d’une fermeture chirurgicale pour éviter la majoration de la valvulopathie aortique.
• Classe IIa : CIV peu restrictives, de taille moyenne, avec symptômes en général discrets (essoufflement pour des efforts importants). En échographie, le gradient transseptal est moyennement élevé (3-4 m/s), l’hyperdébit pulmonaire est net (Qp/Qs > 2, VG dilaté) ; il peut y avoir un gradient pulmonaire modéré d’hyperdébit (VD/AP< 25 mmHg). Ces CIV sont exceptionnellement rencontrées chez l’adulte, car le suivi a été débuté dès l’âge pédiatrique chez ces patients ; elles doivent toutefois être fermées avec, en cas de doute sur le niveau des RVP, réalisation préalable d’un cathétérismes cardiaque.
• Classe IIb : CIV larges non restrictives (> 1 cm) avec HTAP d’hyperdébit chez le nourrisson. Elles ne concernent pas l’adulte. Ces CIV peuvent évoluer vers toutes les autres classes, voire vers une fermeture spontanée.
• Classe III : large CIV non restrictive (> 1 cm) avec HTAP fixée (syndrome d’Eisenmenger). Ici le patient est cyanosé, surtout à l’effort, il n’y a plus de souffle de CIV mais plutôt un B2 fort et un souffle d’IT. Le Doppler sur la CIV montre l’absence de gradient transseptal (Vmax = 1 m/s), le sens du shunt dépend du rapport RVP/RVS. Comme dans tous les syndromes d’Eisenmenger, ces CIV ne doivent pas être fermées, le patient est éligible pour les thérapeutiques usuelles de l’HTAP.
• Classe IV : CIV à « poumons protégés ». En réponse à l’hyperdébit pulmonaire des premiers mois de vie, un obstacle se constitue entre la CIV et l’artère pulmonaire, le plus souvent par prolifération musculaire médioventriculaire droite, parfois par sténose sous-pulmonaire ou valvulaire pulmonaire.
L’hémodynamique est donc celle d’une tétralogie de Fallot. Si le gradient intra-VD ou VD/AP permet d’avoir des pressions pulmonaires basses (Vmax > 4 m/s), ces CIV sont fermées vers l’âge de 18 mois, une fois que la phase de prolifération musculaire médio-VD est stabilisée. Le chirurgien effectuera une fermeture de la CIV par patch et une résection de cette zone hypertrophique, qui le plus souvent est située en région sous infundibulaire. La surveillance consiste ensuite à vérifier l’absence d’hypertrophie résiduelle du VD et de trouble du rythme ou de conduction.
Chez l'adulte, en pratique
La majorité des CIV seront de classe I (maladie de Roger). La surveillance reposera surtout sur l’absence d’apparition de lésions associées, en particulier valvulaires aortiques ; il faut aussi s’assurer que la CIV est vraiment restrictive, avec absence de dilatation des cavités gauches et Qp/Qs à 1.
En l’absence de sténose pulmonaire et aortique, la Vmax sur la CIV devra être directement corrélée à la pression artérielle systémique prise au brassard lors de l’échographie : les PAP systoliques doivent être inférieures au tiers des pressions systémiques ; chez un patient avec une pression artérielle de 120/60 mmHg, les PAP doivent donc être < 40 mmHg (120/3) ; le gradient entre le VG et le VD devra donc être au moins de 80 mmHg (PVGs-PVDs = 120-40 = 80 mmHg), soit une Vmax par la CIV > 4,5 m/s (loi de Bernouilli simplifiée). Ce raisonnement simple n’est valable qu’en l’absence de sténose pulmonaire (PAPs = PVDs) et aortique (PAos = PVGs). Si les PVD sont entre 1/3 et 2/3 des pressions systémiques chez un adulte, la CIV n’est probablement pas suffisamment restrictive : l’indication d’évaluation précise des RVP et de leur vasoréactivité par cathétérisme cardiaque est légitime ; on peut se retrouver alors dans la situation de la « zone grise » préalablement décrite dans les CIA et dont le raisonnement est identique ici.
Chez ce même patient, si le gradient par la CIV est entre 3,5 et 4,5 m/s, avec un VG dilaté (> +2 DS) l’hémodynamique est probablement celle d’une CIV IIa : on calcule alors PAPs = PVDs = 120 - 4 x (3,5)² = 70 mmHg soit PAP/PAo = 0,6.
L’indication de fermeture est à retenir après cathétérisme si les RVP sont basses (HTAP par hyperdébit pulmonaire).
Le cathétérisme permet aussi d’éliminer une autre cause d’hypertension pulmonaire dans ces situations de doute à l’échographie : HTAP postcapillaire associée (dysfonction VG), retour veineux pulmonaire anormal partiel, collatérale systémicopulmonaire, etc. En cas d’HTAP modérée d’origine mixte préet post-capillaire, le calcul des RVP est crucial avant de fermer le shunt. Dans ces situations exceptionnelles, des biopsies pulmonaires peuvent être effectuées pour préciser le niveau d’atteinte vasculaire pulmonaire.
La fermeture des CIV, quel que soit l’âge, reste très majoritairement un geste chirurgical.
Seules les CIV musculaires centrales et certaines CIV apicales en fromage de gruyère peuvent bénéficier d’une fermeture percutanée, comme alternative à la fermeture chirurgicale. La décision doit de toute façon comprendre l’avis du chirurgien, d’autant que certaines procédures actuelles peuvent exceptionnellement associer chirurgie cardiaque et cathétérisme interventionnel (procédures dites « hybrides »). Le positionnement et le largage de la prothèse est effectué sous ETO. La fermeture des CIV périmembraneuses avec une prothèse Amplatzer® adaptée à leur anatomie doit encore être évaluée, mais la majorité des centres ne recommande pas cette technique (zone d’ancrage limitée en zone sous-aortique, proximité des valves et du tissu de conduction).
La tétralogie de Fallot
C’est la cardiopathie cyanogène la plus fréquente, représentant environ 10 % des CC. Arthur Fallot décrit en 1888 les qutre signes caractéristiques : CIV, sténose infundibulaire, dextroposition de l’aorte, hypertrophie ventriculaire droite. En réalité, la déviation antéro-droite du septum conal lors de l’embryogenèse est à elle seule responsable de ces quatre anomalies.
En cas d’association à d’autres anomalies (naissances anormales des coronaires, CIV additionnelles, hypoplasie ou sténose des branches pulmonaires, collatérales aorto-pulmonaires, etc.), on parle de forme irrégulière de tétralogie de Fallot. La forme extrême, où la valve pulmonaire est atrétique, est appelée « atrésie pulmonaire avec CIV». Le diagnostic anténatal est de plus en plus fréquent et permet une naissance en milieu spécialisé (maternités de niveau III avec service de cardiopédiatrie). La cure chirurgicale a lieu vers l’âge de 6 mois sous CEC; elle comprend la fermeture de la CIV par patch et la levée de l’obstacle sous pulmonaire : valvulotomie, résection infundibulaire, patch sur la voie de sortie du VD. Aujourd’hui le patch n’est transannulaire que si l’anneau pulmonaire est hypoplasique et/ou la valvule dysplasique : on a alors une fuite pulmonaire postopératoire obligatoire.
Le patch transannulaire étant majoritairement pratiqué dans le passé, de nombreux patients adultes suivis aujourd’hui ont une fuite pulmonaire souvent marquée.
La surveillance d’un patient avec tétralogie de Fallot opérée est systématique à vie, et en général annuelle. L’ECG analyse la conduction avec mesure de la durée du QRS (facteur de risque de mort subite si QRS > 180 ms). L’échocardiographie vérifie l’absence de shunt résiduel par la CIV, mesure le gradient VD/AP avec si possible analyse des branches pulmonaires (difficile chez l’adulte), contrôle l’absence de valvulopathie aortique (aorte ascendante classiquement dilatée) et étudie la fuite pulmonaire (IP) et la fonction VD.
Les facteurs péjoratifs sont :
PVDs > 60 mmHg, gradient VD/AP > 40m mHg, IP avec surcharge volumique du VD et dysfonction VD.
En cas de fuite pulmonaire significative, les indications de valvulation pulmonaire retenues actuellement sont :
- insuffisance cardiaque,
- diminution de la capacité à l’effort : l’épreuve d’effort, dans l’idéal avec ergospirométrie, permet un suivi longitudinal du patient (VO2 max, seuil ventilatoire),
- arythmies ventriculaires et/ou QRS > 180 ms (stimulation ventriculaire programmée),
- dilatation et/ou dysfonction du VD.
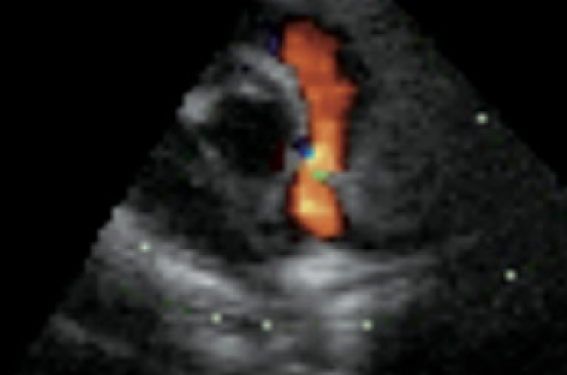
Quantification de la fuite
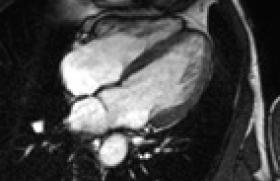
Sauf lorsqu’elle est massive (figure 2), l’analyse et la quantification de la fuite pulmonaire est difficile en échocardiographie, bien que quelques critères soient utiles (rapport VDd/VGd élevé, diamètre à l’origine du flux couleur de l’IP, rapport IP/anneau, IP naissant des branches pulmonaires). À ce jour, il n’y a pas de critères échographiques validés prédictifs d’une dysfonction VD permettant une valvulation plus précoce.
Figure 2. Fuite pulmonaire massive.
L’IRM cardiaque est devenu l’examen de référence avant valvulation pulmonaire, avec un seuil de dilatation du VD de 170 ml/m en diastole. Pour Therrien (Am J Cardiol 2005) et Henkens (Ann Thor Surgery 2007, Int J Cardiol 2007), dans les IP séquellaires d’ancienne tétralogie de Fallot, il existe une diminution du volume du VD après chirurgie réparatrice ; cette récupération postopératoire est moins bonne si l’intervention est tardive, ce qui est le cas de nombreux adultes opérés il y a plus de 20 ans, et bien sûr, si une fuite pulmonaire réapparaît secondairement. La fraction de régurgitation pulmonaire est considérée comme sévère si > 45-50 %, mais il s’agit d’un indicateur mal corrélé à la récupération après remplacement valvulaire pulmonaire. Les indices de volume et la fraction d’éjection corrigée du VD seraient plus pertinents. Pour espérer une retour du VTD en deçà de 108 ml/m2 (ce qui reste encore une valeur élevée puisque l’index de VTD du VD ne devrait pas dépasser 100 ml/m2 chez les sujets normaux), il faudrait intervenir avant que les volumes ventriculaires droits indexés diastoliques et systoliques ne dépassent respectivement 170 et 85 ml/m2. L’IRM constitue donc un guide précieux de quantification dans ce domaine : elle permet de mesurer la fraction de régurgitation pulmonaire par cartographie des flux et celle des volumes ventriculaires droits par contourage.
Intervention
La valvulation chirurgicale comporte le remplacement de la valve pulmonaire par une xénogreffe, comme le conduit type Contegra® (veine jugulaire bovine), par une homogreffe ou encore par une bioprothèse bovine. Le choix du type de valve se fait en fonction de l’âge, des conditions anatomiques, des désirs du patient, de la disponibilité de l’implant, et de la préférence du chirurgien.
Le cathétérisme interventionnel est une nouvelle et prometteuse alternative au traitement chirurgical, par implantation percutanée d’un stent valvé en position pulmonaire (figure 3).
Les premières interventions ont été réalisées à l’Hospital for Sick Children Great Ormond Street à Londres, après l’invention par Philippe Bonhoffer de la valve Melody® (valve biologique fixée à l’intérieur d’un stent). À ce jour, plus de 1 000 patients dans le monde ont bénéficié de cette technique. Le succès de la procédure est très proche de 100 % et les complications rares. Actuellement en France, un STIC est en cours avant commercialisation.
Figure 3. Implantation percutanée de la valve Mélody®.
En pratique
La cardiologie congénitale de l’adulte, avec une prévalence croissante et une population hétérogène, ayant suivi les avancées de la chirurgie cardiaque depuis les années 80, est une jeune « sur-spécialité » de la cardiologie. Elle nécessite des compétences variées : cardiopédiatres, échographistes, rythmologues, chirurgiens, radiologues, cathétériseurs. On a vu ici, par l’illustration de trois cardiopathies apparemment simples et fréquemment rencontrées chez l’adulte, que de nombreuses questions peuvent se poser depuis le diagnostic jusqu’à la thérapeutique.
La cardiologie congénitale est désormais reconnue par le plan maladie rares via la labellisation d’un centre de référence national et de centres de compétence régionaux pour les cardiopathies congénitales complexes. Le manque de formation initiale dans ce domaine reste toutefois un souci pour de nombreux cardiologues.
Très récemment, nous avons déposé un projet de DESC de cardiologie pédiatrique et congénitale : il a été validé par la SFC, par la Société française de pédiatrie, par le Collège des professeurs de cardiologie et de pédiatrie et par le ministère de l’enseignement supérieur et de la recherche. Il est actuellement entre les mains du ministère de la Santé.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
