Grand angle
Publié le 30 mar 2025Lecture 10 min
Prise en charge des malformations vasculaires superficielles - La multidisciplinarité reste la clé
Annouk Anne BISDORFF, Coordinatrice CNMR, Anomalies vasculaires superficielles de l’enfant et de l’adulte, service de neuroradiologue, hôpital Lariboisière, Paris
Les anomalies vasculaires (anciennement appelées « angiomes ») regroupent plusieurs entités classiquement distinguées en deux grandes catégories : les tumeurs et les malformations vasculaires. Les anomalies vasculaires peuvent être rencontrées à tout âge, avec des caractéristiques cliniques, hémodynamiques, histologiques. La prise en charge doit être multidisciplinaire dans la plupart des cas.
Les malformations vasculaires superficielles (extracérébrales) sont des anomalies congénitales, le plus souvent sporadiques, des vaisseaux se manifestant par des lésions artérielles, capillaires, veineuses ou lymphatiques, et peuvent être combinées entre elles.
Les malformations vasculaires sont classées en fonction de la vitesse du flux dans la lésion : flux lent (malformation capillaire, veineuse, lymphatique, ou combinées capillaro-veino-lymphatique) ou flux rapide (malformation artérioveineuse, fistule artérioveineuse).
La complexité de ces entités a été facilitée par l’adoption d’une nomenclature de référence proposée par la Société internationale pour l’étude des anomalies vasculaires (ISSVA)(1).
Poser le diagnostic
La démarche diagnostique est guidée par une corrélation clinique (l’histoire de la maladie et l’examen clinique), et les différentes modalités d’imagerie (échographie Doppler, IRM, TDM) :
- l’âge d’apparition de la lésion ;
- l’histoire de la maladie : apparition à la naissance, progressive ou brutalement après un choc ou des poussées évolutives ;
- l’aspect clinique de la lésion : couleur, chaleur, caractère pulsatile, dépressible, élastique ou molle ;
- l’imagerie qui permet d’aider au diagnostic.
En première intention, l’échoDoppler permet de distinguer les tumeurs vasculaires en différenciant une composante tissulaire des malformations vasculaires soit à flux lent ou flux rapide à l’aide du flux et l’index de résistance. Les imageries en coupes telles que l’IRM et/ou l’angioscanner sont indispensables, afin de déterminer la localisation et l’extension exacte, ainsi que leurs rapports avec les structures avoisinantes et l’envahissement de certaines structures musculaires, osseuses ou nerveuses. Les séquences anatomiques en IRM indispensables sont T1, T2 avec et sans suppression de graisse et parfois T1 avec injection de gadolinium. Les séquences IRM de flux (TRICKS, TWIST) ne sont pas toujours nécessaires, notamment si l’échoDoppler et l’examen clinique ont pu différencier un flux lent d’un flux rapide. L’artériographie est indiquée que si un éventuel traitement endovasculaire ou chirurgical d’une malformation artérioveineuse est prévu, mais pas à titre diagnostique sauf exception. Là encore, la cartographie Doppler éventuellement des IRM dynamiques ou des angioscanners et des reconstructions 3D peut être utile et sont moins invasive.
Les malformations artérioveineuses
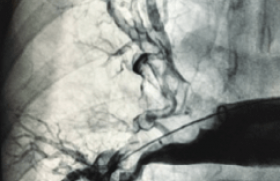
Une malformation artérioveineuse (MAV) se caractérise par une communication anormale entre des artères et des veines sans lit capillaire, ceci génère des flux rapides. Il n’existe pas de lésion tissulaire, ni de lit capillaire (figure 1), ce qui diminue la résistance et augmente ainsi la vitesse à travers les shunts artérioveineux (shunts AV). Les vaisseaux afférents s’allongement et deviennent tortueux (figure 2).
Figure 1. A. Représentation schématique normale : entre une artérielle afférente (rouge), lit capillaire et retour veineux (bleu). B. Malformation artérioveineuse avec une artère afférente dilatée, cercle : nidus avec nombreux shunt artérioveineux (*) et drainage veineux avec un retour veineux (dilation de la veine efférente). C. Vue en imagerie.
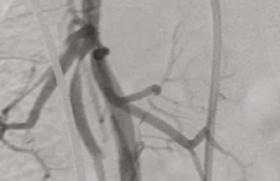
Figure 2. A. Malformation artérioveineuse (syndrome de Parkes Weber) de la jambe, avec plusieurs points de shunt AV. B. ÉchoDoppler : shunt artérioveineux : aliazing et flux systodiastolique, avec baisse de l’index de résistance.
Les MAV sont congénitales et bénignes. Elles apparaissent au début de la période de gestation(1,2). Souvent cliniquement invisibles dans la petite enfance, ces lésions grandissent proportionnellement avec le patient et deviennent symptomatiques qu’après la puberté. Les changements hormonaux au cours de la puberté, contraception et/ou au cours de la grossesse peuvent déclencher la croissance de ces malformations vasculaires. Les traumatismes corporels, y compris les interventions telles que des embolisations proximales ou des chirurgies partielles, peuvent également déclencher une progression en induisant une augmentation des facteurs de croissance des vaisseaux sanguins.
La plupart des MAV sont sporadiques et des études récentes ont révélé certaines mutations génétiques(3,4). Il existe de rares formes familiales de MAV(5), généralement associées à d’autres anomalies dans un contexte syndromique, telles que la télangiectasie hémorragique héréditaire (HHT ou maladie de Rendu-Osler), le syndrome de malformation capillaire-malformation artérioveineuse (CM-AVM)(6) et le syndrome de Cowden (PTEN)(7). Ces maladies sont dues à des mutations germinales, c’est-à-dire que le gène responsable de la malformation a été hérité de l’un ou des deux parents.
La symptomatologie des MAV peut être variable. La tuméfaction associée ou non à une pulsatilité peuvent entraîner une gêne et une douleur. Parfois, la destruction tissulaire adjacente secondaire à une surcharge veineuse entraîne une ulcération cutanée, des épisodes hémorragiques et, exceptionnellement, une insuffisance cardiaque à haut débit. La classification clinique de Schobinger permet d’évaluer l’évolution du patient et poser une éventuelle indication thérapeutique.
Les avancées en génétique, des dix dernières années, ont permis dans les tumeurs vasculaires et malformations vasculaires superficielles de mettre en évidence des mutations germinales et somatiques sporadiques de gènes impliqués dans les voies de signalisation cellulaire. Les principales voies de signalisation impliquées sont :
- PI3K /AKT/mTOR : différentes kinases activées en cascade(8) ;
- RAS/RAF/MEK/ERK, mettant en jeu des récepteurs membranaires à activité tyrosine kinase ;
- VEGF A (facteur de croissance vasculaire endothéliale) A/VEGFR2 (récepteur du VEGF).
Quelle prise en charge ?
La prise en charge thérapeutique(9) est guidée par la symptomatologie clinique et le souhait du patient. Les questions auxquelles il faut répondre sont les suivantes : qui doit être traité et pourquoi ? Comment traiter la MAV ? Quand traiter et par qui ? Les principales options thérapeutiques sont soit la gestion du traitement médical (suivi clinique et vêtements de compression, bêtabloquants, thérapie médicale ciblée), soit des traitements plus invasifs tels que l’embolisation endovasculaire/la ponction directe et/ou l’excision chirurgicale de la MAV. Elle doit être discutée avec le patient en lui expliquant le bénéfice et les risques des différentes attitudes thérapeutiques après une concertation multidisciplinaire. En fonction de l’âge du patient, de la localisation de la lésion, du type agiographique, la stratégie thérapeutique peut se décliner en fonction des stades cliniques (tableau) :
- stade de Schobinger I (dormance sans symptôme) : le traitement est conservateur. En l’absence de gêne clinique, une surveillance régulière et des mesures préventives, telles qu’éviter les facteurs d’aggravation (traumatismes, traitement hormonal) avec le port d’une compression élastique est indispensable quand la localisation le permet. Elle permet une réduction du débit (pour éviter les complications cutanées veineuses dues à l’hypertension veineuse) et également la protection de la MAV des traumatismes ; parfois un traitement médicamenteux peut être associé, comme des bêtabloquants (ceci permet de diminuer la surcharge vasculaire et la pulsatilité) ;
- stade Schobinger II-III (expansion-nécrose) : les stades supérieurs à II présentant une progression clinique et/ou des épisodes hémorragiques récurrents peuvent bénéficier soit d’un traitement médical, soit de techniques d’embolisation ciblées et/ou d’une chirurgie d’exérèse en fonction de la localisation de la MAV, de sa taille et de ses caractéristiques d’imagerie. Un traitement immunosuppresseurs dans les stades III, peut être proposé du fait de leurs propriétés antiangiogéniques (sirolimus, thalidomide, etc.)(10,11), celui-ci précédé parfois les traitement invasifs (chirurgie ou embolisation) ;
- dans certains rares cas avec une mutation Map2K1, des thérapies ciblées (anti-MEK)(12) et/ou Kras(13) ont également été proposées.
Et la radiologie interventionnelle
Les techniques de radiologie interventionnelle ont dominé le traitement des MAV au cours des 20 dernières années(14,15). Les progrès techniques dans les approches artérielles, veineuses et vasculaires percutanées, l’amélioration du matériel de cathétérisme endovasculaire et des agents emboliques ont permis de réduire le nombre d’échecs post-traitement. Le nombre de séances de traitement a été réduit.
L’analyse précise de l’architecture des vaisseaux de la MAV joue un rôle important dans la décision de traitement.
La classification de Yakes(16) (figure 3) peut être utile dans la prise de décision.
Figure 3. Classification de Yakes. Type 1 : FAV directe. Type 2a : MAV artérioveineuse + nidus + veines. Type 2b : 2a + s’abouchant veine de drainage unique anévrismale. Type 3a : multiples artères + artérioles + veine drainage unique. Type 3b : multiples artères dans une veine de drainage unique puis multiples veines. Type 4 : multiples artères et artérioles en passage et microfistules dans multiples veines.
Il est très important de ne pas procéder à une ligature chirurgicale ou à une embolisation proximale par coils/plugs des artères afférentes alimentant la MAV. C’est une contre-indication absolue si l’on n’est pas en mesure d’occlure la totalité du nidus de la MAV.
La plupart des MAV sont sporadiques et des études récentes ont commencé à élucider la base génétique de ces lésions avec l’identification de variants somatiques dans les gènes de la voie de signalisation RAS/MAPK (figure 4).
Figure 4. Schéma de la voie de signalisation RAS/MAPKinas.
Les gènes MAP2K1 et KRAS sont retrouvés chez plus de 60 % des patients. La plupart de ces mutations sont également identifiées dans des tumeurs malignes où elles jouent un rôle « moteur » pour la croissance tumorale et pour lesquelles des thérapies ciblées (inhibiteurs de MEK) ont déjà été développées et largement utilisées en oncologie. Ces thérapies ciblées pourraient offrir, étant donné la similarité des cibles, une nouvelle stratégie thérapeutique pour les cliniciens qui prennent en charge les patients atteints de MAV. Deux études de cas récentes ont montré l’efficacité d’un inhibiteur de MEK (tramétinib)(17) dans les MAV extracrâniennes. Lekwuttikarn(18) et coll. ont rapporté un cas de MAV dorsale extra crânienne mutée MAP2K1 chez une patiente de 11 ans, et Edwards et coll.(19) ont décrit l’impact du tramétinib dans la prise en charge d’une forme syndromique de MAV avec une mutation KRAS.
Dans certaines études pilotes, l’effet antiangiogénique de la thalidomide a montré une amélioration clinique chez les patients traités pour épistaxis chez les patients HHT et d’autres équipes l’ont utilisé comme une utilisation hors étiquette dans le traitement de la MAV superficielle(20).
Plus récemment, Laurence Boon et coll. ont publié une étude de cas très intéressante sur le traitement à la thalidomide chez des patients atteints de malformations artérioveineuses graves(21).
Dans les malformations à flux lent (veineuses ou lymphatiques)
L’utilisation du sirolimus(21,22) est devenue une norme de soins pour les malformations lymphatiques et veineuses, bien que les anomalies vasculaires ne soient pas encore une indication approuvée pour ce dernier. Des études de phase 3 sont en cours(23).
• Malformations veineuses
• La prise en charge médicale, notamment par compression élastique, est un volet important. Elle permet de diminuer la distension veineuse et la douleur, et de réduire également le risque de thrombose intramalformatif.
• L’abstention thérapeutique est privilégiée pour les malformations veineuses de petite taille, peu symptomatiques dans les topographies non à risque et s’il n’y a pas de gêne fonctionnelle ni de demande esthétique du patient. Une surveillance clinique annuelle reste justifiée.
• Les traitements curatifs discutés sont :
– l’exérèse chirurgicale ;
– les traitements endoveineux : sclérothérapie échoguidée et sous scopie ou laser thermique endoveineux ; cryothérapie ou radiofréquence peuvent être proposés ;
– le sirolimus (rapamycine) peut être indiqué en traitement adjuvant à la chirurgie, afin de prévenir le risque de complications thrombotiques et hémorragiques.
• Malformations lymphatiques (MLK)
La stratégie thérapeutique est discutée en concertation pluridisciplinaire en différenciant la forme macrokystique de la forme micro - kystique, et en prenant en compte l’âge du patient, l’extension de la MLK et sa localisation. En l’absence de gêne fonctionnelle, l’abstention thérapeutique avec suivi régulier est proposée. La physiothérapie associant compression adaptée et ajustée à la croissance de l’enfant est proposée en fonction de la localisation.
En cas de nécessité de traitement de fond, l’objectif est de maintenir la fonctionnalité et de limiter l’impact esthétique en prévenant l’aggravation de la MLK. Les différentes modalités thérapeutiques peuvent être combinées entre elles, répétées au cours du suivi avec des périodes d’abstention thérapeutique(24) :
– la sclérothérapie sous contrôle scopique représente le traitement de première intention de la plupart des MLK, plus efficace dans les formes macrokystiques que dans les MLK microkystiques. Elle est réalisée à distance des épisodes infectieux ;
– d’autres traitements comme électrocoagulation, laser NYAG, laser endovasculaire, etc., sont utiles pour détruire les vésicules superficielles, les lésions microkystiques et pour tarir les épisodes de saignements. Ils sont utilisés dans les MLK avec lymphangiectasies cutanées ou des muqueuses(25) ;
– la prise en charge chirurgicale est discutée en deuxième intention. À l’exception des petites malformations lymphatiques bien circonscrites, la résection ne peut être que partielle dans la plupart des MLK en raison de l’infiltration en profondeur. Il s’agit d’une chirurgie de réduction dans les lésions déformantes avec gêne fonctionnelle, ou dans les formes tissulaires ;
– les médicaments immunosuppresseurs, inhibiteurs de mTOR(26), interfèrent avec la voie de signalisation PI3K-AKT-mTOR et agissent en inhibant la lymphangiogenèse. Ils sont utilisés depuis 2011 dans les MLK compliquées. Il s’agit d’un traitement suspensif, efficace sur les symptômes douloureux, lymphorrhée et microhémorragies. Le sirolimus (rapamycine)(24), d’administration orale, est le plus ancien et le plus utilisé en traitement de fond pour les MLK. L’alpélisib semble également prometteur dans cette indication chez des patients mutés Pik3 CA(27). Une surveillance clinique et biologique régulière tous les 2-3 mois est requise : risque infectieux, de cytopénies, concentration résiduelle du médicament, risque d’interaction avec les inhibiteurs ou les inducteurs du CYP3A4.
Conclusion
La démarche diagnostique pour préciser le type de malformation (flux lent, flux rapide ou syndrome combiné), est guidée par une corrélation radioclinique. La prise en charge thérapeutique doit être discutée pour la plupart des cas lors d’une concertation multidisciplinaire. Cette prise en charge devient de plus en plus multimodale : surveillance clinique, traitement médicamenteux et/ou plus invasif : radiologie interventionnelle et/ou exérèse chirurgicale.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :