

Publié le 18 jan 2015Lecture 14 min
Intérêt de la génétique dans la mort subite sur cœur apparemment sain
F. SACHER, Hôpital cardiologique du Haut-Lévêque, université Bordeaux-II, C. ROORYCK-THAMBO, Service de génétique médicale, CHU de Bordeaux
L’intérêt de la génétique dans la mort subite familiale ne se résume pas à la recherche d’une mutation. Elle permet l’identification et la prévention des personnes à risque dans la famille. Pour cela, la cause de la mort subite doit être recherchée, une enquête familiale réalisée et enfin pour certaines pathologies on cherchera une mutation génétique.
Cette mise au point se limite aux morts subites rythmiques sur cœur sain.
Rechercher l’étiologie de la mort subite
Il s’agit d’une étape essentielle pour la suite de la prise en charge car les causes de mort subite sont multiples, et loin d’être toutes héréditaires (infarctus du myocarde, embolie pulmonaire, rupture d’anévrisme...). De plus, l’identification de l’étiologie permet une meilleure acceptation du décès, et ce qui était vécu comme une fatalité devient la conséquence d’une pathologie.
Si le patient est décédé, il faut récupérer le compte rendu d’autopsie, et demander du matériel biologique s’il est disponible (parfois possible si des prélèvements toxicologiques ont été réalisés par la justice et si le décès est très récent) pouvant servir à une analyse génétique ultérieure. Les circonstances du décès sont également primordiales à recueillir (âge lors du décès < 45 ans, circonstances de survenue - repos, sommeil, effort -, douleurs thoraciques avant le décès, facteurs de risque cardiovasculaire, autres morts subites dans la famille, âge lors de ces morts subites). Tous ces éléments permettront d’orienter le diagnostic, donc de proposer une enquête familiale appropriée.
S’il s’agit d’un patient avec mort subite récupérée, les choses sont plus « simples », et le patient bénéficie d’un bilan complet : interrogatoire à la recherche des mêmes éléments que ci-dessus pour orienter le diagnostic, ECG, Holter-ECG longue durée, échocardiographie et épreuve d’effort. Puis, en fonction du contexte et des premiers résultats, on pourra réaliser des tests pharmacologiques (ajmaline, adrénaline, iso-prénaline), une coronarographie et une IRM cardiaque (forme fruste DAVD, CMH apicale, non-compaction). La recherche d’une mutation génétique n’est réalisée que lorsqu’un diagnostic permettant d’orienter l’analyse génétique est posé.
Enquête familiale
Elle est indispensable en cas de mort subite (récupérée ou non) liée à une pathologie rythmique héréditaire afin de prévenir un autre décès dans la famille. Les formes familiales identifiées sont principalement de transmission autosomique dominante (avec des sujets, hommes ou femmes, atteints à chaque génération - transmission dite « verticale »), mais il existe aussi des formes de transmission autosomique récessive (apparition de la pathologie dans une génération donnée chez plusieurs descendants, transmission dite « latérale » dans l’arbre généalogique). Dans le cas d’une transmission autosomique dominante, le dépistage familial doit commencer par les apparentés ascendants et descendants du 1er degré du cas index(1) (père, mère, et enfants en fonction de leur âge et de la pathologie identifiée), puis en fonction des résultats obtenus chez les parents du cas index, le dépistage se poursuit chez les frères, sœurs, oncles et tantes du cas index, et continue de proche en proche (figure 1). Autant l’enquête familiale est souvent réalisée lorsque le patient survit et est hospitalisé en cardiologie autant lorsque le décès survient en dehors de l’hôpital voire dans un service de réanimation médicale non sensibilisé à ce problème, elle est rarement proposée à la famille.
Arbre généalogique
Figure 1. Patient de 27 ans (III-1) asymptomatique jouant en fédérale 1 de rugby vu pour la visite médicale habituelle de début de saison. Le médecin est interpellé par la mort subite de son grand-père à 41 ans et par le fait que son père et 2 de ses oncles sont implantés d’un défibrillateur pour une myocardiopathie dilatée à coronaires saines. En poussant plus loin les investigations, le diagnostic de laminopathie avec mutation dans le gène de la lamine A/C sera porté chez eux et la mutation sera également retrouvée chez le sujet III-1.
Absence de pathologie identifiée
Si aucune pathologie n’a pu être identifiée chez la personne décédée, mais qu’on pense à une pathologie héréditaire, alors les circonstances de survenue du décès sont essentielles à faire préciser (cf. chapitre précédent). Pour l’apparenté venant consulter, on commence toujours par un interrogatoire à la recherche de syncopes ou équivalents (respiration agonique nocturne, crise tonico-clonique...). On pratique ensuite un ECG 12 dérivations en continu avec manœuvres de Valsalva, puis en remontant les électrodes de 2 espaces intercostaux (V1 et V2 au 2e espace intercostal) pour améliorer la détection d’un syndrome de Brugada(2). En fonction du contexte clinique (cf. infra - description des différentes pathologies rythmiques héréditaires) et de l’aspect ECG, on oriente les examens. Toutefois, le bilan de première intention comprend généralement une échocardiographie (bien que très rarement contributive dans ce contexte), un Holter-ECG (pour identifier la survenue de trouble du rythme - exceptionnel -, mais surtout les changements morphologiques de QRS et de longueur de QT, en particulier la nuit) et une épreuve d’effort (d’autant plus intéressante que les morts subites familiales surviennent dans un contexte adrénergique ou en cas de suspicion de QT long - non-raccourcissement à l’effort). Ensuite, en fonction des données recueillies, on peut proposer un test aux bloqueurs des canaux sodiques (ajmaline, flécaïne, procaïnamide) si l’aspect ECG est suspect de Brugada ; un test à l’isoprénaline(3), si contexte catécholergique ; un test à l’adrénaline(4,5) (figure 2), si suspicion de QT long ; une IRM si suspicion de DAVD ou de myocardiopathie débutante.
Figure 2. Protocole pour la réalisation du test à l’adrénaline en cas de suspicion de QT long et d’intervalle QT limite.
Syndrome du QT long
Il s’agit d’une entité regroupant des pathologies différentes (> 10 formes) ayant en commun un allongement de l’intervalle QT corrigé supérieur à 440 ms chez les hommes et 460 ms chez les femmes.
Les trois premières formes décrites de ce syndrome représentent la grande majorité des formes retrouvées (70-80 %).
Le QT long de type 1 (environ 40 % des syndromes du QT long) est caractérisé par une onde T large avec une pente ascendante lente et surtout par la survenue fréquente de syncope à l’effort, tout particulièrement en nageant. Toute syncope en piscine doit faire évoquer la possibilité d’un QT long. Il répond particulièrement aux bêtabloquants qui permettent de diminuer très nettement le risque de mort subite. Il est rare de devoir recourir à l’implantation d’un défibrillateur. Le dépistage familial dans cette forme requiert outre l’ECG, un Holter-ECG (variations de la longueur du QT sur les 24 heures) et une épreuve d’effort (absence de raccourcissement du QT à l’effort). En cas de doute persistant, on peut réaliser un test à l’adrénaline (figure 3) qui permet de porter le diagnostic avec une sensibilité de 91 % et une spécificité de 100 %(4). Il est dû à une mutation dans le gène KCNQ1 entraînant une perte de fonction du canal potassique lent (Iks).
Le QT long de type 2 (environ 30 % des syndromes du QT long) est caractérisé par une onde T bibosse. Les arythmies ventriculaires surviennent préférentiellement lors de stress émotionnel ou auditif (réveil, sonnerie de téléphone), mais parfois également à l’effort. Le traitement bêtabloquant est un peu moins efficace que pour le QT long de type 1, et il faut parfois recourir à l’implantation d’un défibrillateur automatique implantable (DAI). Les mêmes examens que pour le QT long de type 1 doivent être réalisés avec un intérêt particulier pour le Holter-ECG car la durée du QT est souvent variable au cours du temps. Il est dû à une mutation dans le gène KCNH2 entraînant une perte de fonction du canal potassique rapide (Ikr).
Le QT long de type 3 représente environ 10 % des syndromes du QT long, mais cette forme possède le risque rythmique le plus élevé. La mort subite est fréquemment le mode d’entrée dans la maladie. Les arythmies ne semblent pas liées à une décharge catécholergique, et l’efficacité des bêtabloquants est controversée chez des sujets qui sont, en plus, souvent bradycardes à l’état basal. Il est fréquent de devoir implanter un DAI. Cette forme est due à une mutation dans le gène SCN5A entraînant un gain de fonction du canal sodique entrant.
Le syndrome du QT long de type 7 (syndrome d’Andersen-Tawil) est rare, mais il a des caractéristiques très particulières. Il associe une dysmorphie faciale (hypoplasie mandibulaire, hypertélorisme, oreilles bas implantées, petite taille et parfois clinodactylie et syndactylie des doigts et/ou des orteils), une paralysie périodique (douleur musculaire à type de crampes par moment) et des signes ECG caractéristiques. Le QT est long, mais aux dépens d’une onde U proéminente (aspect pseudo-bibosse), par ailleurs il existe des ESV polymorphes fréquentes et parfois des TV bidirectionnelles (figure 3) relativement lentes et bien tolérées. Le risque de mort subite semble relativement faible chez les patients sous bêtabloquants, même s’ils ne font pas disparaître les troubles du rythme ventriculaire. Cette forme est due à une mutation dans le gène KCNJ2 dans la majorité des cas qui entraîne une perte de fonction du canal potassique Kir 2.1.
Figure 3. TV bidirectionnelle (astérisques) quasi permanente chez un enfant de 9 ans asymptomatique atteint d’un syndrome de QT long de type 7 (Andersen-Tawil). Il est parfois difficile d’observer l’intervalle QT et l’onde T car les arythmies sont très fréquentes. Il existe parfois un bigéminisme polymorphe. La morphologie de l’onde T est très particulière avec un aspect de pseudo-bibosse souvent liée à une onde U proéminente (flèche).
Tachycardies ventriculaires catécholergiques
Elles se caractérisent par un risque élevé de mort subite chez les sujets jeunes typiquement à l’effort, les bêtabloquants représentent donc le traitement de choix au long cours(6). Elles sont parfois confondues avec une épilepsie chez l’enfant, ce qui peut être dramatique. L’ECG de base est normal, mais la survenue de troubles du rythme ventriculaire bidirectionnels et polymorphes à l’effort est caractéristique, même si à l’âge adulte ces troubles du rythme peuvent être moins marqués. L’épreuve d’effort est donc l’examen clé dans cette pathologie. Les mutations décrites impliquent le gène du récepteur à la ryanodine 2 (RYR 2) ou de la calséquestrine (CASQ 2) qui jouent un rôle dans le métabolisme calcique.
Syndrome de Brugada
Le syndrome de Brugada, caractérisé par un sus-décalage du segment ST dans les dérivations précordiales droites, est maintenant bien connu de tous les cardiologues. Le risque rythmique de cette pathologie apparaît finalement assez faible chez les sujets asymptomatiques (0,5 % par an versus 0,1 % par an dans la population générale). Le bilan familial en cas de mort subite liée à un syndrome de Brugada commence par un interrogatoire à la recherche de syncope, puis la réalisation d’un ECG standard, puis avec V1 et V2 au 2e espace intercostal ce qui permet de le sensibiliser. Si l’aspect est douteux, on propose un test de provocation avec l’injection d’un bloqueur des canaux sodiques (ajmaline, flécaïne, procaïnamide). Dans le cadre d’un bilan familial, si l’ECG est strictement normal, on discute au cas par cas la réalisation de ce test de provocation. En effet, chez les sujets asymptomatiques en l’absence d’aspect de type 1 spontané, que le test soit positif ou négatif, cela ne change pas forcément l’attitude thérapeutique, en revanche, cela peut avoir un impact sur la suite de l’enquête familiale.
Une mutation du gène SCN5A codant pour le canal sodique est retrouvée chez environ 20 % des patients présentant un syndrome de Brugada. Toutefois, de plus en plus d’éléments vont contre une transmission monogénique liée à SCN5A(7) ce qui rend le conseil génétique complexe et la recherche de la mutation moins utile en pratique clinique. Par ailleurs, des mutations sur d’autres gènes ont été décrites, mais restent anecdotiques.
Syndrome de repolarisation précoce
Il s’agit d’un syndrome décrit récemment, associant une élévation du point J d’au mois 0,1 mV dans au moins 2 dérivations, en particulier les dérivations inférieures (figure 4), à l’exception de V1 à V3, à la survenue de fibrillations ventriculaires(8). La survenue de ces FV n’a pas de lien avec l’effort, et cette entité partage des similitudes ECG avec le syndrome de Brugada (syndromes d’élévation du point J). Le traitement après mort subite récupérée est bien sûr le DAI, mais en cas d’orage rythmique, l’isoprénaline est efficace en aigu, tandis que la quinidine l’est en chronique. Il n’existe pas encore assez de données pour préconiser une attitude particulière chez les sujets asymptomatiques, mais la hauteur de l’élévation du point J semble corrélée à la récidive des arythmies ventriculaires. Des mutations ont été décrites sur différents gènes (KCNJ8, CACNA1C, CACNB2b), mais pour l’instant, cela reste du domaine de la recherche.
Figure 4. Jeune homme de 24 ans avec mort subite ressuscitée dont l’ECG à distance retrouve un aspect compatible avec un syndrome de repolarisation précoce (élévation du point J de 0,2 mV à type de crochetage dans les 3 dérivations inférieures). Le bilan, comprenant les tests pharmacologiques, l’IRM cardiaque et la coronarographie avec Méthergin, sera negative.
Laminopathies
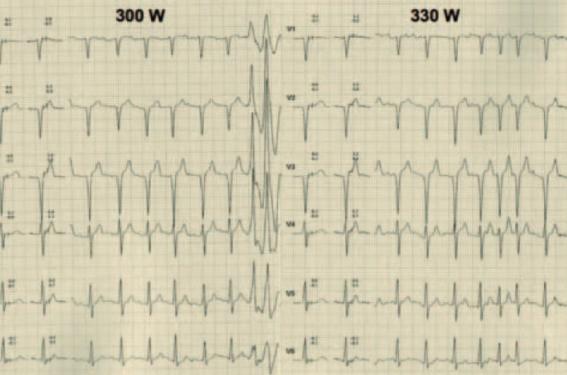
Elles n’entrent pas à proprement parler dans le cadre des pathologies responsables de mort subite sur cœur sain, car les patients développent des myocardiopathies dilatées à coronaires saines. Toutefois, la mort subite peut survenir assez tôt dans l’évolution de la myocardiopathie. Il s’agit d’une pathologie rare, mais trop souvent méconnue par les cardiologues. Elle associe une symptomatologie musculaire (faiblesse et atrophie des muscles de la région humérale et péronéale, rétraction tendineuse) et une symptomatologie cardiaque. Le piège est que parfois seule la symptomatologie cardiaque est présente. Sur le plan cardiaque, cela commence typiquement vers 20 ans avec des troubles du rythme atriaux, puis des troubles de la conduction AV (seulement PR long au début) (figure 5a et b). Puis vers 40 ans, il existe un risque important de mort subite. Ceux qui ne décèdent pas évoluent vers l’insuffisance cardiaque avec développement d’une myocardiopathie dilatée à coronaires saines.
Le diagnostic et le bilan familial sont d’autant plus importants que la transmission est autosomique dominante (1 enfant sur 2 sera atteint) et qu’un test génétique est disponible (recherche de mutation dans le gène LMNA codant pour la lamine A/C). Les patients mutés ont une indication de DAI (même si le moment de l’implantation n’est pas définitivement tranché).
Figure 5a. Patient III-1 de la figure 1. Son ECG de base retrouve une bradycardie sinusale, un BAV 1, un QRS anormal avec un aspect QS de V1 à V3 et une ESV infundibulaire sans caractère de malignité.
Figure 5b. À l’épreuve d’effort, il atteint 330 W. On note un doublet ventriculaire venant du ventricule gauche (tracé de gauche) avec 3 morphologies d’ESV isolées (non montrées) et des salves d’arythmie atriale (tracé de droite). Par ailleurs, on observe que le PR reste long à l’effort, ce qui est pathologique. L’IRM montrera des zones de rehaussement tardif limitées signant une myocardiopathie débutante. Les analyses génétiques seront réalisées et retrouveront la mutation familiale. Il sera alors interdit de sport et implanté d’un DAI.
Tests génétiques
La réalisation des tests génétiques obéit à plusieurs règles :
1. Le dépistage familial ne peut être réalisé que si la mutation causale est identifiée dans un gène connu comme étant impliqué dans la pathologie. En effet, les gènes sont différents d’une pathologie à l’autre, et il n’est pas envisageable en pratique clinique d’étudier tous les gènes pour une question de coût, de temps, mais surtout de pertinence clinique. Ainsi lorsqu’on va « à la pêche », on a très peu de chance de trouver une mutation connue comme étant pathogène, en revanche, on peut trouver des variants nucléotidiques (modification rare d’une base dans un gène dont l’effet sur la fonction du gène n’est pas avéré et donc la signification clinique non connue) qui vont, au final, compliquer l’explication donnée au patient.
2. Le patient doit être informé de la réalisation de ce test et des conséquences éventuelles, il doit signer un consentement éclairé.
3. Chez les patients non atteints cliniquement - phénotype négatif - (QT normal dans une famille de QT long...), les tests sont encore plus réglementés. En effet, on parle alors de dépistage génétique présymptomatique, réalisé au sein d’une équipe pluridisciplinaire déclarée au ministère. Ces tests ne se conçoivent que si une mutation clairement pathogène a été identifiée dans la famille. Ils sont interdits chez les mineurs s’il n’existe pas de bénéfice thérapeutique ou préventif à brève échéance.
4. Le prélèvement génétique lui-même est très simple puisqu’il s’agit d’une prise de sang dans un tube à NFS qu’il faut envoyer (avec les documents cliniques et réglementaires) au centre faisant l’analyse. Ces analyses s’effectuent généralement dans un centre dit « de référence » qui choisit en fonction des données cliniques et électrocardiographiques fournies le ou les gène(s) à analyser. Ces analyses prennent en moyenne de 6 mois à 1 an. Lorsqu’on connaît déjà la mutation familiale, seule cette mutation est recherchée. Les résultats sont donc plus rapides (dans le mois). Une fois la mutation trouvée, elle doit être confirmée sur un second prélèvement sanguin ou par frottis jugal.
5. Les résultats doivent être rendus par le médecin prescripteur de l’analyse génétique, lors d’une consultation médicale individuelle. Récemment, des centres dits de « compétence » en lien avec des centres de référence ont été créés pour gérer cela. En pratique, il existe des consultations conjointes avec cardiologue et médecin généticien, parfois psychologue, afin de délivrer une information complète au patient sur les résultats génétiques et le dépistage familial à effectuer. Il est parfois difficile de comprendre qu’on est atteint d’une pathologie, mais qu’aucune mutation n’ait été retrouvée. De même, la différence entre variant et mutation est parfois difficile à appréhender. Enfin, la recherche de mutations sur un gène peut être négative alors même que le gène est anormal. L’analyse du gène est réalisée par « zone d’intérêt » (régions codantes essentiellement), mais elle n’est pas exhaustive ; de plus, avec les techniques stan- dards, on peut passer à côté de délétions ou duplications dans ces mêmes gènes (9).
Comme tout autre examen, les tests génétiques n’ont ni une sensibilité (la non-détection d’anomalie du gène est possible) ni une spécificité (variant nucléotidique sans conséquence clinique) de 100 %. Enfin, il est important d’expliquer le mode de transmission de la pathologie en cause pour compléter l’enquête familiale et répondre aux questions des parents concernant leurs enfants ou une éventuelle grossesse.
En conclusion
La recherche d’une mutation génétique dans un bilan de mort subite familiale intervient après la mise en évidence d’une pathologie rythmique héréditaire due à un ou des gènes identifiés. Lorsque la mutation d’un gène est retrouvée, cela permet d’aider l’enquête familiale. Mais cette dernière est, de toute façon, indispensable (qu’une mutation soit identifiée ou pas) afin de prévenir une mort subite chez les apparentés.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
