




Publié le 14 fév 2015Lecture 9 min
Atteinte polyartérielle au cours de maladies vasculaires rares
T. MIRAULT, J. EMMERICH, E. MESSAS, Hôpital européen Georges-Pompidou, Paris
À l’instar de l’athérosclérose, certaines pathologies artérielles rares peuvent affecter les troncs supra-aortiques, les coronaires, l’aorte, les artères viscérales et les artères des membres inférieurs. Il s’agit de pathologies constitutives de la paroi artérielle, comme les élastopathies, ou de pathologies inflammatoires de la paroi artérielle, comme les vascularites. Voici un aperçu de trois maladies vasculaires rares : la dysplasie fibromusculaire, la maladie de Takayasu et la maladie de Horton.
La dysplasie fibromusculaire
Le terme de dysplasie fibromusculaire (DFM) s’applique à un groupe de maladies idiopathiques, segmentaires, non inflammatoires et non athéroscléreuses de la paroi artérielle, entraînant des sténoses des artères de petit et moyen calibres. La DFM est une maladie rare dont les patients présentent pour principaux signes ou symptômes une HTA et/ou des manifestations neurologiques. Pour le diagnostic, on utilise actuellement la classification radiologique fondée sur des corrélations anatomopathologiques. On distingue ainsi principalement trois formes de DFM :
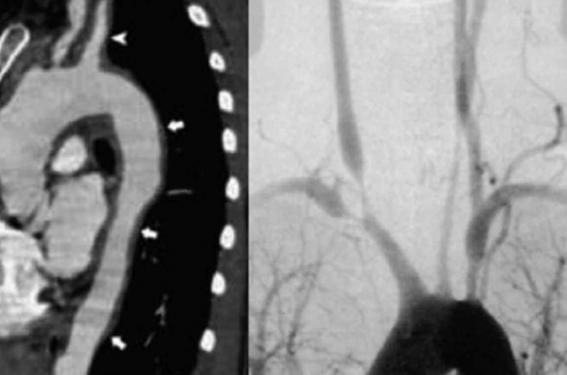
- la DFM médiale (60-70 %), dont l’aspect angiographique typique est une image en « perles enfilées » par succession de sténoses et d’anévrismes (figure 1) ; ces aspects correspondent histologiquement à des zones d’hypertrophie fibreuse de la média et de rupture de la limitante élastique interne ; ce type de DFM, le plus fréquent, s’observe principalement vers la quarantaine ;
- la DFM périmédiale ou sous-adventitielle (10-20 %), dont l’aspect angiographique est celui d’une sténose dysplasique tubulaire ; histologiquement, les couches externes de la média sont le siège d’une hyperplasie de la matrice extracellulaire refoulant l’adventice et réduisant la lumière du vaisseau ;
- la DFM intimale (5 %) qui se présente angiographiquement comme une sténose unifocale ; histologiquement, elle est caractérisée par un épaississement intimal circonférentiel sans atteinte des autres tuniques.
Figure 1. Atteinte carotidienne avec aspect de perles enfilées au cours de la dysplasie fibromusculaire (DFM).
La DFM peut être familiale : la survenue de DFM rénale a été en effet décrite chez des paires de jumeaux identiques et dans des fratries. Par écho-tracking à haute résolution, des anomalies asymptomatiques et non sténosantes des carotides communes ont été trouvées chez les parents du premier degré des cas index.
Atteinte des artères rénales
L’atteinte des artères rénales est présente chez environ 60 % des patients atteints de DFM. La présentation symptomatique de la DFM des artères rénales est une hypertension artérielle (HTA) rénovasculaire. La prévalence de cette présentation est estimée à 10 % des cas d’HTA rénovasculaire et à moins de 4/1 000 dans la population hypertendue d’âge moyen (soit approximativement 8 nouveaux cas pour 100 000 personnes actives et par an).
Atteinte des troncs supra-aortiques (TSA)
La DFM des TSA peut se manifester par des acouphènes pulsatiles et être responsable d’accident vasculaire cérébral ischémique (avec ou sans dissection) ou plus rarement hémorragique par rupture d’anévrisme intracrânien. La prévalence des lésions symptomatiques de DFM des TSA reste mal connue.
Atteinte d’autres territoires
Les sténoses dysplasiques du territoire mésentérique peuvent entraîner des nausées, des douleurs digestives et une perte de poids. Ces présentations sont plus souvent associées à l’athérosclérose. En l’absence d’argument pour l’athérosclérose (sujet jeune sans facteurs de risque d’athérosclérose et n’ayant pas de plaque d’athérome), il est logique de rechercher des lésions de DFM. La DFM des artères mésentériques, sous-clavières et aorto-iliaques peut être associée à une ischémie dans ces territoires. Les explorations étiologiques réalisées dans ces situations permettent le diagnostic de DFM.
Prise en charge
Le traitement de l’HTA rénovasculaire repose sur un traitement antihypertenseur, l’angioplastie transluminale percutanée (ATP) des sténoses fonctionnelles et une reconstruction chirurgicale dans les cas complexes où les artères segmentaires sont atteintes. Les options thérapeutiques pour les lésions dysplasiques des TSA incluent un traitement médical (antithrombotiques essentiellement) et très rarement un traitement endovasculaire ou chirurgical. La DFM n’est pas une maladie endothéliale : l’indication des statines ne diffère pas des indications usuelles en cas d’hypercholestérolémie ou de maladie athéroscléreuse associées.
La maladie de Takayasu
Parmi les vascularites des vaisseaux de gros calibre, on retrouve la maladie de Horton, et la maladie de Takayasu. Ces deux artérites granulomateuses affectent l’aorte et ses principales branches de division (figure 2). La maladie de Takayasu, encore appelée « maladie des femmes sans pouls » ou syndrome de Martorell est universellement répandue et affecte la femme (70 % des cas) jeune (30 ans). La physiopathologie reste obscure, probablement multifactorielle : certaines séries retrouvent plus de 50 % d’antécédent tuberculeux, mais rarement de tuberculose maladie patente au diagnostic, de multiples associations à des affections auto-inflammatoires, auto-immunes sont rapportées : spondylarthrite ankylosante, maladie de Crohn, polyarthrite rhumatoïde, lupus érythémateux systémique, etc. Sur le plan histologique, les lésions sont multifocales, segmentaires, prédominant à la jonction médio-adventitielle, à type de sclérose dense du collagène, d’une fragmentation de la média d’un infiltrat inflammatoire lympho-plasmocytaire et de quelques cellules géantes. Le diagnostic repose sur un faisceau d’arguments cliniques, radiologiques et biologiques, chez une femme jeune bien souvent, parfois complété d’arguments histologiques.
Figure 2. Atteinte de l’arche aortique avec sténoses des troncs supra-aortiques au cours de la maladie de Takayasu.
Manifestations générales
Fièvre, altération de l’état général accompagnent la phase inaugurale de la maladie. S’y associent des manifestations articulaires à type d’arthralgies, et parfois arthrites, des myalgies, des sérites, un érythème noueux. Lors des poussées de la maladie, un syndrome inflammatoire biologique est volontiers observé : accélération de la vitesse de sédimentation, élévation moins fréquente de la CRP, hypergammaglobulinémie.
Atteinte vasculaire
Phénomène de Raynaud, claudication des membres supérieurs ou inférieurs, angor mésentérique, carotidodynies, HTA, amauroses fugaces, éclipses, angor coronaire, dyspnée, amènent à la découverte d’abolition de pouls, d’une asymétrie tensionnelle et surtout de souffles cervicaux, sous-claviers, abdominaux ou lombaires. Ces symptômes sont secondaires aux manifestations ischémiques engendrées par les lésions : succession de sténoses localisées et d’ectasies, occlusions, anévrismes sacculaires. Les territoires les plus souvent rencontrés sont les carotides primitives, les sous-clavières postvertébrales, la crosse aortique, l’aorte thoracique descendante ou abdominale, les artères viscérales, les artères pulmonaires. L’artériographie met en évidence des sténoses longues effilées, régulières, et des dilatations artérielles, alors que l’écho-Doppler et l’imagerie en coupes (scanner et IRM) retrouvent en plus des disparités de calibre, l’épaississement homogène, circonférentiel, avec prise de contraste tardive de la paroi artérielle, évocateur d’artérite inflammatoire. Le TEP-scan détecte par ailleurs le caractère hypermétabolique de ces épaississements pariétaux.
Atteinte cardiaque
coronarite ostiale est responsable d’une symptomatologie d’insuffisance coronarienne dans près de 10 % des observations. Les atteintes valvulaires sont rares, mais peuvent occasionner une insuffisance aortique, une insuffisance mitrale.
Une hypertension artérielle pulmonaire peut témoigner d’une atteinte des artères pulmonaires présente dans 50 % des cas de maladie de Takayasu.
Prise en charge
La corticothérapie est préconisée dans les formes évolutives, en favorisant la régression de la symptomatologie clinique, la réapparition des pouls, la normalisation du syndrome inflammatoire biologique. Habituellement débutée à 1 mg/kg/jour d’équivalent prednisone, elle est progressivement diminuée dès le 1er mois. La récidive des symptômes ou l’aggravation des sténoses lors de la décroissance cortisonique peut amener à intensifier le traitement par ajout d’un immunosuppresseur : méthotrexate, voire d’un anti-TNF : infliximab. Le contrôle tensionnel est majeur mais rendu difficile, d’une part, par les difficultés de mesures (prise systématique de la pression aux 4 membres) et, d’autre part, par les symptômes cérébraux ou oculaires secondaires à une hypotension en aval des sténoses carotidiennes par exemple. La levée d’une coarctation aortique ou d’une sténose artérielle rénale, digestive, sous-clavière ou des membres inférieurs peut améliorer la symptomatologie et aider au contrôle tensionnel. Que ce soit par chirurgie à ciel ouvert ou par voie endovasculaire, on essaiera dans la mesure du possible d’être à distance de toute poussée et sous une corticothérapie faible. Enfin, l’arrêt du tabac est recommandé, et une statine fréquemment prescrite.
La maladie de Horton
Parmi les vascularites des vaisseaux de gros calibre, on retrouve la maladie de Horton, et la maladie de Takayasu. La maladie de Horton survient chez la personne de plus de 50 ans avec une incidence estimée à 20/100 000 par an. La maladie s’installe en général progressivement, avec céphalées, altération de l’état général, manifestations rhumatismales, parallèlement à un syndrome inflammatoire biologique. Le diagnostic repose sur des arguments cliniques et biologiques, et si possible une confirmation histologique. Les lésions vasculaires sont segmentaires et focales en histologie, avec inflammation granulomateuse avec cellules géantes multinucléées au sein de la média, rompant la limitante élastique interne. L’intima peut être le siège d’une prolifération fibreuse restreignant la lumière et favorisant les thromboses (figure 3).
Figure 3. Atteinte sténosante de l’artère fémorale superficielle gauche, occlusion de l’artère fémorale superficielle droite, au cours de la maladie de Horton.
Manifestations générales
La fièvre est retrouvée dans plus de 50 % des cas, de simples fébricules à une véritable fièvre hectique ou en plateau. L’altération de l’état général est variable, parfois prononcée dans les formes pseudo-néoplasiques.
Atteinte articulaire
Inconstantes, les manifestations articulaires associent arthralgies de la ceinture scapulaire ou de la ceinture pelvienne, d’horaire inflammatoire, enraidissement du rachis cervical, arthralgies inflammatoires des grosses articulations plus rarement. Le tableau évoque une pseudo-polyarthrite inflammatoire.
Atteinte vasculaire
Les céphalées, typiquement temporales, témoignent d’une atteinte du territoire carotidien externe, et sont le signe le plus constant et le plus évocateur de la maladie de Horton. Des céphalées occipitales, une claudication à la mastication, des dysesthésies du cuir chevelu sont des symptômes classiquement rapportés bien qu’inconstants. L’examen clinique retrouve une induration des artères temporales, voire une abolition du pouls temporal. Des souffles cervicaux, sous-claviers peuvent être retrouvés. L’écho-Doppler artériel, l’angioscanner, l’angio-IRM, non nécessaires au diagnostic, mettent en évidence des sténoses longues effilées, régulières, par épaississement homogène, circonférentiel, avec prise de contraste tardive de la paroi artérielle ou halo hypoéchogène, évocateur d’artérite inflammatoire. Le TEP-scan détecte par ailleurs le caractère hypermétabolique de ces épaississements pariétaux. Le territoire le plus souvent atteint est le territoire carotidien externe (95 % dans une série autopsique) : artères temporales, faciales, occipitales. La crosse de l’aorte peut être atteinte (20-40 %) avec épaississement pariétal, anévrisme, dissection, et révèle parfois la maladie. Dans 10 à 15 % des cas, une atteinte artérielle des membres supérieurs et/ou inférieurs est rapportée, se manifestant par une claudication, des ischémies digitales, des troubles trophiques. Enfin, de rares cas de coronarite ostiale sont rapportés, généralement associés à une aortite.
Atteinte ophtalmique
Les complications oculaires sont graves et représentent le risque évolutif majeur de la maladie. Ces complications sont de nature ischémique : neuropathie ischémique antérieure aiguë, neuropathie optique rétrobulbaire aiguë, occlusion de l’artère centrale de la rétine. La présence de signes d’alerte : diplopie transitoire, amaurose, limitation douloureuse des mouvements oculaires, ne doit pas retarder l’instauration de la corticothérapie et d’un antiagrégant plaquettaire.
Prise en charge
La corticothérapie évite la survenue de complications oculaires et permet une sédation des douleurs, une apyrexie, et une amélioration de l’état général. Habituellement débutée à 0,7 mg/kg/jour d’équivalent prednisone, la corticothérapie est progressivement diminuée dès le 1er mois. Le sevrage se fera prudemment, les rechutes sont fréquentes, 30 à 40 %. La durée de la corticothérapie sera bien souvent supérieure à un an. La prévention de l’ostéoporose est très importante dans cette population âgée. La récidive des symptômes ou l’aggravation des sténoses lors de la décroissance cortisonique peut amener à intensifier le traitement par ajout d’un immunosuppresseur : le méthotrexate.
En pratique
Une atteinte polyartérielle est classiquement retrouvée chez les patients atteints d’athérosclérose.
D’autres pathologies vasculaires, beaucoup plus rares que l’athérosclérose, atteignent les vaisseaux de gros et moyen calibres.
Une des missions du plan maladies rares 2004 est de sensibiliser les étudiants en médecine à ces maladies.
Cet article brosse un aperçu de ces trois maladies vasculaires rares, que tout médecin peut un jour être amené à suivre et à diagnostiquer devant un cas d’atteinte polyartérielle, comme le résume le tableau ci-dessous.
Pour en savoir plus
Dysplasie fibromusculaire
• Dysplasie fibromusculaire symptomatique chez l’adulte. PNDS ALD hors liste de la HAS - www.has-sante.fr
• Plouin PF et al. Orphanet J Rare Dis 2007 ; 2 : 28.
• Touze E et al. Int J Stroke 2010 ; 5 : 296-305.
• Slovut DP, Olin JW. N Engl J Med 2004 ; 350 : 1 862-71.
• Trinquart L et al. Hypertension 2010 ; 56 : 525-32.
Maladie de Takayasu
• Arnaud L et al. Medicine (Baltimore) 2010 ; 89 : 1-17.
• Arnaud L et al. Rev Med Interne 2009 ; 30 : 578-84.
• Fiessinger JN. Ann Med Interne (Paris) 1994 ; 145 : 538-40.
• Numano F et al. Lancet 2000 ; 356 : 1 023-5.
Maladie de Horton
• Melson MR et al. Rev Neurol Dis 2007 ; 4 : 128-42.
• Fraser JA et al. Rev Neurol Dis 2008 ; 5 : 140-52.
• Assie C et al. Medicine 2011 ; 90 : 40-51.
• Assie C, Marie I. Presse Med 2011 ; 40 : 151-61.
• Le Hello C et al. J Rheumatol 2001 ; 28 : 1 407-12.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
