




Publié le 15 sep 2020Lecture 4 min
Mort subite et calcification artérielle généralisée chez l’enfant
Sonia SALMI-BELMIHOUB, Philippe CHEVALIER, service de rythmologie, hôpital cardiologique Louis Pradel, Lyon
La calcification artérielle généralisée de l’enfant (GACI : generalized arterial calcification of infancy) est une maladie génétique caractérisée par une calcification extensive des artères de moyen et gros calibre. Elle se traduit par des sténoses artérielles et une ischémie myocardique sévère avec une mortalité infantile importante. Nous décrivons le cas d’une enfant de 13 ans dont l’histoire clinique a débuté à l’âge de 26 mois dans ce contexte de GACI.
A.H., née le 25 septembre 2006, présente à l’âge de 26 mois dans un contexte d’hypertension artérielle un oedème aigu du poumon compliqué de choc cardiogénique. Le scanner thoraco-abdominal montre des calcifications artérielles périphériques au niveau des reins, de la rate et du pancréas faisant évoquer un tableau de GACI.
La fraction de raccourcissement est calculée alors à 26 %. Un traitement médical est débuté.
Le bilan génétique découvre une mutation ABCC6 hétérozygote. L’évolution de la maladie est émaillée par une mort subite en 2014 par fibrillation ventriculaire récupérée après 3 chocs électriques externes. Le traitement par digoxine initialement prescrit est remplacé par amiodarone. L’indication d’un défibrillateur est retenue mais non implanté. La patiente est régulièrement suivie, un contrôle angiographique coronaire en 2017 montre des sténoses très étendues du réseau gauche ainsi qu’une lésion infiltrative très serrée de la coronaire droite distale avec atteinte de l’IVP et la RVP avec une suppléance du réseau gauche par des branches septales issues de la coronaire droite. Il existe aussi une sténose très serrée de la sousclavière gauche, une sténose modérée de la sous-clavière droite, de la vertébrale droite, absence de sténoses sur les artères rénales, la fraction d’éjection ventriculaire gauche échographique est calculée à 25 % (figure 1).
Figure 1. (A) Lésions étendues du réseau gauche. (B) Sténose sous-clavière gauche (à gauche) ; sténose de la croix du cœur, coronaire droite (à droite).
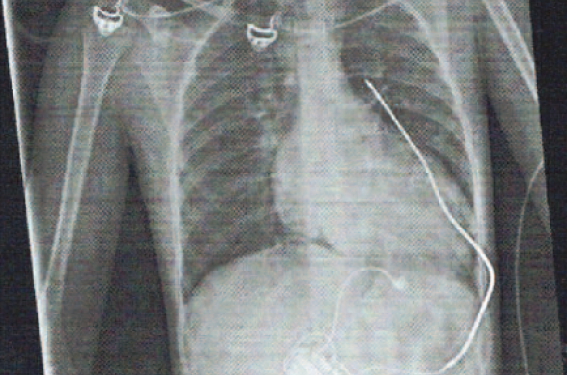
Un défibrillateur implantable est mis en place par voie chirurgicale en mars 2018 avec sonde ventriculaire droite de détection stimulation épicardique et électrode de défibrillation sous-cutanée, le boîtier est intra-abdominal.
A.H. est hospitalisée en juillet 2019 pour syncope correspondant à une fibrillation ventriculaire traitée par choc électrique interne. Trois chocs électriques de 35 J ont été nécessaires pour un retour au rythme sinusal. Le traitement médical est optimisé avec majoration des bêtabloquants et dose de charge d’amiodarone.
Après 9 mois de suivi, il n’y a pas eu de récidive de troubles rythmiques et l’examen clinique est stable sans complications extracardiaques liées à l’amiodarone (figures 2 et 3).
Figure 2. Fibrillation ventriculaire traitée par CEI.
Figure 3. DAI.
Discussion
La calcification artérielle généralisée de l’enfant est une maladie génétique rare, grave, sa prévalence est estimée à 1 pour 391 000 naissances(1). Elle est caractérisée par l’installation dès la phase in utero de calcifications intimales des artères de moyen et gros calibre(2). La majorité des patients survivants après la naissance développent des symptômes cardio-pulmonaires significatifs(2). Une complication rythmologique (fibrillation ventriculaire) a déjà été décrite dans un contexte de syndrome coronaire aigu chez un adolescent de 16 ans(3).
Le pédiatre peut être alerté par des chiffres tensionnels élevés une insuffisance rénale, une insuffisance cardiaque. D’autres signes extracardiaques peuvent survenir à un âge plus avancé, comme des calcifications articulaires, une fragilité osseuse, un rachitisme, une perte de l’audition, des papules jaunâtres au niveau des aisselles, des plis inguinaux, plis de flexion du genou et des coudes, et des stries angioïdes à l’examen du fond d’œil, un certain nombre de ces symptômes sont similaires à ceux observés dans le pseudo-xanthome élastique (PXE), maladie du métabolisme calcique d’origine génétique.
Bien que le diagnostic puisse être réalisé in utero par échographie anténatale, il est le plus souvent fait tôt après la naissance, devant des symptômes tels que : insuffisance cardiaque, détresse respiratoire, hypertension artérielle, cardiomégalie, hémorragie par rupture d’artères calcifiées. Le scanner et l’IRM sont recommandés pour quantifier les calcifications.
Physiopathologie
Il s’agit d’une accumulation de calcium qui débute durant la période foetale avec diminution du pyrophosphate à l’origine d’un dépôt exagéré de calcium dans les vaisseaux. Le pyrophosphate prévient la calcification en adhérant aux cristaux minéraux et en augmentant l’ostéopontine (figure 4).
Figure 4. Calcification artérielle et pyrophosphate.
Il existe deux formes génétiques autosomales récessives de GACI.
La plus fréquente est due à une mutation du gène ENPP1 qui code pour une enzyme nécessaire à la formation de pyrophosphate dont le taux diminue, favorisant les calcifications artérielles. Le type 2 est dû à une mutation du gène ABCC6.
La majorité des enfants et adultes atteints de la mutation du gène ABCC6 ne développent pas de GACI, mais présentent une pathologie dite pseudo-xanthome élastique atteignant le tissu élastique de la peau, des yeux, du système cardiovasculaire et gastro-intestinal, ces calcifications surviennent plutôt chez le grand enfant et l’adulte.
Traitement
Une thérapie par biphosphonates analogues du pyrophosphate utilisés dans le traitement de l’ostéoporose est proposée le plus tôt possible afin de tenter de réduire l’accumulation tissulaire calcique. Un traitement prénatal peut être débuté si le diagnostic est fait in utero. Le pronostic de cette pathologie demeure grave, la mortalité est lourde, notamment les 6 premiers mois de vie.
L’atteinte rythmologique chez notre patiente ne semble pas spécifique du GACI. Il correspond à une complication de l’atteinte artérielle coronaire.
En pratique
La calcification artérielle généralisée de l’enfant est une maladie génétique rare, mais grave à début précoce in utero dont le diagnostic est fait souvent à un stade avancé de la maladie (insuffisance cardiaque, insuffisance rénale).
L’amélioration des techniques de dépistage in utero pourrait permettre un traitement précoce en période anténatale et donc d’améliorer le pronostic de cette pathologie.
La biologie moléculaire et l’identification de l’une ou de l’autre des formes ENPP1 ou ABCC6 sont utiles.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
