


Publié le 10 nov 2009Lecture 7 min
Cardiomyopathie d'allure « idiopatique » : quel bilan étiologique ?
F. BAUVAIS, Département de Cardiologie, Hôpital Lariboisière
Les Journées françaises de l’insuffisance cardiaque
Quand et pourquoi adresser le patient en consultation génétique ?
P. CHARRON (Paris)
La cardiomyopathie dilatée (CMD) est la plus fréquente des cardiomyopathies. La génétique moléculaire a permis des progrès considérables dans la compréhension de cette pathologie. Selon les séries, il apparaît que 20 à 35 % sont des formes familiales dont la transmission est le plus souvent autosomique dominante. En fait, les formes génétiques sont très hétérogènes, les mutations identifiées touchant des gènes codant pour des protéines diverses de la structure et du fonctionnement de la cellule myocardique.
La consultation génétique permet d’établir ou de rectifier un diagnostic, d’identifier un sous-type de CMD et d’aider au diagnostic prédictif chez les apparentés. L’impact de cette consultation sur les modalités de suivi, les choix thérapeutiques et la détermination du risque familial permet d’améliorer significativement la prise en charge du patient atteint et de sa famille (figure 1).
Figure 1. Impact du dépistage génétique.
Un certain nombre de cas cliniques présentés durant cette session ont permis d’appréhender différents concepts :
• Concept de remodelage avec l’âge : un diagnostic de CMD porté à 50 ans peut en fait révéler être une forme évoluée de CMH familiale.
• Concept de variabilité phénotypique dans une même famille : dans le cadre du bilan familial d’une CMD, d’autres formes de cardiomyopathies peuvent être diagnostiquées (figure 2).
• Concept de diagnostic précoce pour les formes génétiques les plus graves : les laminopathies sont des affections génétiques hétérogènes compliquées de CMD à haut risque de mort subite rythmique chez l’adulte jeune, d’où la nécessité d’un diagnostic précoce conduisant à un bilan rythmologique invasif systématique pour discuter la mise en place d’un défibrillateur.
Figure 2. Cas clinique de variabilité phénotypique.
Lors d’une consultation génétique, la recherche d’un phénotype particulier chez le patient atteint (BAV, myopathie) est systématique.
L’interrogatoire est ciblé sur l’histoire familiale à la recherche d’une CMD, d’une autre cardiomyopathie, d’une mort subite inexpliquée ou d’un phénotype associé (BAV, myopathie). Le bilan familial initial est organisé quel que soit le contexte familial.
Les apparentés ne peuvent pas être directement contactés par l’équipe médicale mais seulement par le patient lui-même. Lors du screening des apparentés, il a été établi que le nombre de réponses est significativement amélioré lorsque l’information est délivrée par courrier plutôt que par voix orale. Un ECG et une échocardiographie sont systématiquement effectués. Le recours au test génétique est proposé chez les apparentés d’un patient atteint dont le gène a été identifié. Le rendement de ce test est faible en cas de CMD commune, forte en cas de CMD avec phénotype associé. Il doit, bien sûr, être effectué en respect des textes de loi actuellement en vigueur. La surveillance des apparentés est poursuivie même en cas de normalité du bilan initial.
Faut-il traquer les virus et lesquels ?
J.-P. GUEFFET (Nantes)
La myocardite virale est une cause de cardiomyopathie connue depuis plus de 50 ans. L’impact de la persistance du génome viral dans la progression de la maladie reste controversé et il n’y a actuellement aucun traitement efficace permettant d’enrayer cette évolution.
Parmi les myocardites infectieuses, les myocardites virales sont les plus fréquentes. De nombreux virus ont été mis en cause, au premier rang desquels les adénovirus, les entérovirus, le parvovirus B19 et les virus coxsakie A et B. L’évolution vers la CMD fait intervenir trois phases successives : réplication virale(1), réponse immune(2), dysfonction myocytaire(3). Il existe probablement une susceptibilité génétique et des facteurs acquis favorisant le développement vers la CMD. Le déficit en sélénium, la grossesse et la malnutrition ont été incriminés.
Le diagnostic de myocardite aiguë n’est pas toujours aisé et repose sur un faisceau d’arguments. Le tableau clinique n’est pas spécifique et peut prendre différentes formes : insuffisance cardiaque aiguë, symptomatologie d’infarctus, troubles du rythme. L’échocardiographie et la biologie sont peu spécifiques.
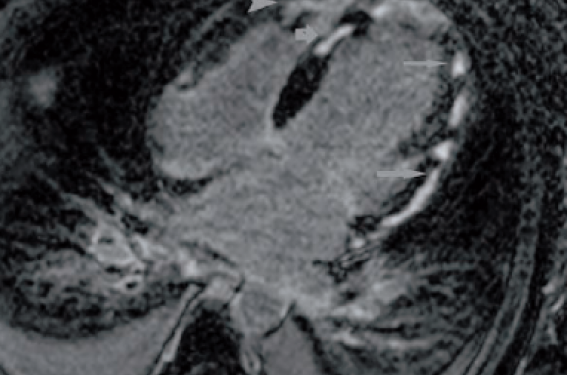
Figure 3. Nodules sous-épicardiques à l’IRM (D’après Chatterjee, Crit Care Med, 2008).
L’IRM est essentielle au diagnostic avec mise en évidence de nodules diffus plutôt superficiels et sous-épicardiques (figure 3). La biopsie endomyocardique (BME) fait le diagnostic mais sa sensibilité n’est que de 60 à 70 % et, devant le risque de complications de ce geste, son indication n’est posée que dans des cas particuliers. Le traitement immunosuppresseur peut se justifier dans le cas de myocardites spécifiques.
Retrouver une étiologie virale au stade de CMD est encore plus difficile. La prévalence d’infections virales et de génome viral retrouvés à la BME est importante chez les sujets présentant une dysfonction ventriculaire gauche « idiopathique » (38 % chez les sujets aux antécédents de myocardite et 20 % chez les patients sans antécédent), mais cette prévalence est également importante dans la population sans dysfonction ventriculaire, ce qui ne permet pas de conclure. D’après Kindermann, le pronostic des patients atteints de CMD dont l’étiologie virale est suspectée dépend surtout de la classe NYHA et de la présence de signes inflammatoires à l’immunohistologie et non de la classification de Dallas à l’histologie ou de la détection du génome viral.
Compte tenu de la physiopathologie de la cardiomyopathie virale, on peut logiquement discuter de traitements spécifiques en fonction du stade de la maladie : traitements antiviraux en phase 1 et traitements immunosuppresseurs en phase 2 et 3. Plusieurs stratégies sont proposées : IL-2 dans les infections à coxsackie B3, interféron ‚ pour éradiquer le génome viral au sein du myocarde, stéroïdes et aziathioprine, agissant sur la phase 2 inflammatoire, et immunoglobulines, essentiellement actives sur la phase 3 avec régulation de l’activité des cytokines.
En pratique
La physiopathologie des CMD d’origine virale est encore mal élucidée. Le diagnostic reste difficile et repose essentiellement sur l’IRM associée ou non à la BEM avec PCR et immunohistochimie.
Le traitement spécifique présente un intérêt pronostique potentiel et l’efficacité des stratégies proposées est actuellement en cours d’étude.
Quand et comment rechercher une pathologie musculaire ?
D. DUBOC (Paris)
Un grand nombre de maladies neuromusculaires ont une atteinte cardiaque associée. Ces pathologies d’origine génétique nécessitent, surtout dans les formes à phénotype variable ou l’atteinte cardiaque peut être la seule manifestation de la maladie, un bilan familial spécifique associant Holter-ECG, échocardiographie, éventuellement IRM et test génétique. Ces bilans familiaux doivent être systématiques quand un des membres d’une famille est atteint.
Quand aucun membre de la famille n’est atteint, le diagnostic de cardiomyopathie d’origine neuromusculaire est plus difficile. La présence de symptômes et de signes cliniques évocateurs (fatigabilité et douleurs musculaires) doit faire évoquer le diagnostic. Les examens complémentaires sont ciblés en fonction des éléments retrouvés à l’interrogatoire et à l’examen clinique : épreuve d’effort avec dosages des lactates, électromyogramme, bilans biologiques et métaboliques spécifiques et biopsie musculaire avec analyses moléculaires et génétiques.
Les laminopathies sont constituées d’un ensemble très hétérogène de pathologies en rapport avec la mutation d’un gène codant pour les lamines A et C. L’atteinte cardiaque conditionne le pronostic vital. Les formes cliniques les plus fréquentes sont une CMD isolée ou associée à une atteinte musculaire périphérique.
Devant un tableau de CMD, les éléments devant faire suspecter une laminopathie sont la présence d’un contexte familial de CMD ou de mort subite, la survenue précoce de troubles du rythme supraventriculaires et conductifs, et l’existence d’anomalies neuromusculaires associées, mêmes minimes.
Une prise en charge spécifique est nécessaire, reposant sur un diagnostic génétique et de larges indications à la mise en place d’un défibrillateur implantable.
Les causes inflammatoires à ne pas méconnaître
G. GRATEAU (Paris)
Dans l’amylose, tous les organes peuvent être potentiellement atteints. Le retentissement clinique et échocardiographique d’une éventuelle atteinte myocardique est systématiquement évalué. À l’inverse, devant un tableau d’insuffisance cardiaque, certains aspects échocardiographiques, essentiellement l’hypertrophie septale, doivent faire évoquer le diagnostic d’amylose. En effet, certaines formes d’amylose ont un pronostic sévère, nécessitant un traitement spécifique adapté afin d’améliorer le pronostic de ces patients. La présence d’anomalies extra cardiologiques, en particulier cutanées, associées au tableau d’insuffisance cardiaque doit être systématiquement recherchée. Le diagnostic d’amylose est confirmé par la biopsie qui peut être effectuée sur la graisse abdominale (AGA) ou les glandes salivaires (BGSA). L’immunohistochimie et la recherche de mutations permettent de déterminer le type de l’amylose (tableau).
Tableau. Les différents types d’amylose.
En pratique
Plusieurs cas cliniques ont été présentés à titre d’exemples :
- Cas clinique 1 : homme de 55 ans présentant une dyspnée progressivement croissante. À l’échocardiographie, présence d’une hypertrophie septale à 17 mm associée à une fraction d’éjection ventriculaire à 45 %. À l’examen clinique, présence de lésions cutanées évocatrices. Diagnostic d’amylose AL dont le pronostic reste sombre en l’absence de traitement adapté.
- Cas clinique 2 : homme de 76 ans, Malien, hospitalisé pour insuffisance cardiaque. Présence d’une hypertrophie septale à l’échocardiographie. Aucune notion d’hypertension artérielle. Diagnostic d’amylose génétique africaine (TTR+ à l’immunohistochimie et mutation V122I).
- Cas clinique 3 : homme de 78 ans. Tableau d’insuffisance cardiaque et de cardiopathie hypertrophique. Canal carpien opéré 4 ans auparavant et présence connue d’une IgG monoclonale. Diagnostic d’amylose sénile.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
