


Publié le 07 juin 2005Lecture 18 min
Myocardiopathie hyperthrophique de l'enfant
D. SIDI, hôpital Necker, Paris
Les myocardiopathies hypertrophiques (MCPH) chez l’enfant recouvrent une diversité sémiologique, échographique et étiologique beaucoup plus importante que celle de l’adulte. En pédiatrie, surtout chez le nouveau-né et le nourrisson, il faut mettre l’accent sur l’enquête étiologique. Ces patients doivent être pris en charge dans des centres très spécialisés avec des compétences non seulement cardiopédiatriques mais aussi génétiques et métaboliques disposant d’un plateau technique et de laboratoires appropriés.
Dans cet article destiné aux cardiologues, nous avons, par un bref rappel embryologique, histologique et physiopathologique du myocarde en formation, tenté d’expliquer les particularités pédiatriques. La conduite à tenir est ensuite traitée en fonction de l’âge sans rentrer dans le détail qui relèverait de la surspécialité.
Pour le lecteur pressé
La plupart des MCPH de l’adulte sont souvent asymétriques, exposent avant tout au risque de mort subite et sont le plus souvent dues à un déficit constitutionnel d’une protéine contractile du myocarde qui se transmet de façon autosomique dominante.
La différence entre MCPH de l’adulte et de l’enfant tient à ce que la plupart des MCP hypertrophiques primitives apparemment isolées du jeune enfant :
• ne survivent pas ;
• se sont intégrées secondairement, avec l’apparition de signes extracardiaques, dans un syndrome génétique polymalformatif, myopathique, neurologique ou métabolique particulier ;
• se sont transformées en MCP dilatées en raison d’une altération sévère de la fonction systolique, ce qui aboutit à une dilatation du ventricule et à un amincissement des parois (la masse se répartissant sur un volume plus important) ;
• ont guéri spontanément sans que l’on sache souvent pourquoi.
En pédiatrie, et surtout chez le nourisson, il faut mettre l’accent sur l’enquête étiologique. Cette enquête s’efforcera avant tout de ne pas méconnaître les MCPH secondaires, les seules accessibles à un traitement spécifique, mais aussi à essayer à tout prix de définir l’origine des MCPH primitives. En effet, trouver la cause des MCPH est essentiel pour le pronostic, pour l’indication ou la contre-indication à une transplantation cardiaque, et le conseil génétique concernant les très nombreux cas de décès de MCPH à court terme.
L’enquête étiologique repose sur l’enquête familiale, la recherche d’anomalies extracardiologiques, souvent discrètes, voire infracliniques, et lorsqu’elles sont présentes, orientent vers des syndromes génétiques polymalformatifs ou métaboliques dont le pronostic neurologique ou musculaire contre-indiquera éventuellement une transplantation cardiaque (TC). En l’absence de diagnostic, on ira jusqu’à la biopsie endomyocardique. Ces patients doivent alors être pris en charge dans des centres très spécialisés avec des compétences non seulement cardiopédiatriques mais aussi génétiques et métaboliques disposant d’un plateau technique et de laboratoires appropriés.
MCPH de l’enfant et de l’adulte : deux approches radicalement différente
MCPH de l’adulte
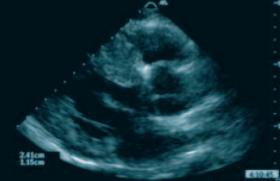
La myocardiopathie hypertrophique est une entité bien définie, due — on le sait depuis maintenant 15 ans — à des anomalies constitutionnelles des protéines contractiles du myocarde qui se transmettent de façon autosomique dominante et se traduisent par une hypertrophie myocardique (par adaptation, la quantité remplaçant la qualité) (figure 1). Le pronostic de ces MCPH est très variable, dépendant des différentes variantes (degré d’hypertrophie, obstruction, trouble du rythme, dysfonction diastolique et éventuellement systolique du myocarde), dont certaines sont corrélées avec le génotype. Les principaux centres d’intérêt dans la recherche clinique sur les MCPH de l’adulte se concentrent sur l’évaluation du risque en fonction des différentes formes échographiques et/ou génotypiques (corrélation phénotype/génotype), la valeur prédictive de certains examens (épreuve d’effort, holter, électrophysiologie) et l’évaluation de différentes formes de traitement classiques ou modernes visant à prévenir ou retarder les complications et, en particulier, la mort subite. Définir les meilleures indications du défibrillateur, du pacing pour remodeler le ventricule, d’une levée de l’obstruction sous-aortique par embolisation ou chirurgie et, enfin, de la transplantation cardiaque.
Figure 1. Hypertrophie ventriculaire gauche de la paroi du septum.
MCPH de l’enfant
Au contraire, les MCPH de l’enfant recouvrent des étiologies et des physiopathologies beaucoup plus diverses, avec des profils évolutifs variables, bien qu’en règle très défavorables. Si cette diversité n’existe plus à l’âge adulte, c’est que la plupart de ces patients ont soit guéri, soit sont décédés, soit enfin se sont intégrés dans des chapitres autres de la pathologie, car les signes extracardiologiques sont apparus et ont pris le pas sur le cœur (myopathie, neuropathie, syndrome génétique polymalformatif ou métabolique).
La difficulté chez l’enfant tient à ce que les formes qui associent la MCPH à des signes extracardiologiques apparaissent souvent au départ sous la forme de MCPH apparemment isolées. Il est important d’en faire le diagnostic étiologique précoce pour le conseil génétique et pour contre-indiquer éventuellement une transplantation cardiaque, si le pronostic neurologique est péjoratif.
Définitions
Embryologie
Le myocarde est un dérivé du mésoderme. Il s’installe dès les premières semaines de la vie embryonnaire et c’est lui qui assure la circulation fœtale. Au début de la grossesse, les cellules sont relativement lâches avec un aspect de myocarde spongieux, dit non-compacté, relativement pauvre en myofibrilles et riche en fibres conjonctives (figure 2). En cours de gestation, et en particulier pendant le troisième trimestre, les myocytes se divisent très activement, les myofibrilles s’organisent architecturalement en parallèle autour du cytosquelette avec une maturation du capital enzymatique de la fibre myocardique et des canaux ioniques, une transformation de la myosine et une multiplication des récepteurs sympathiques myocardiques. Cet ensemble se traduit par une amélioration de la contractilité, de la relaxation et de la compliance du cœur fœtal en fin de grossesse comme s’il se préparait au stress des premiers jours de vie.
Figure 2. Myocarde spongieux non-compacté.
Les cellules myocardiques continuent de se diviser en période néonatale (hyperplasie myocardique avec un énorme potentiel de croissance de la masse myocardique). Au-delà de cette période (quelques semaines), la masse myocardique augmente beaucoup plus lentement et par hypertrophie. La même observation peut être faite pour les capillaires coronaires, si bien qu’avec le temps, la réserve coronaire par gramme de myocarde diminue et qu’une trop forte hypertrophie s’accompagne d’une baisse de la réserve coronaire.
Histologie
Le myocarde est composé de myocytes constitués de filaments d’actine et de myosine qui sont, avec la troponine et la tropomyosine, les principales protéines engagées dans la contraction de la fibre. La dystrophine maintient l’architecture du myocyte et fait partie du cytosquelette. Les cellules myocardiques sont extrêmement riches en mitochondries et le myocarde est l’organe qui consomme le plus d’oxygène par gramme de tissu. Le métabolisme fœtal dépend des sucres et de l’acide lactique sans utilisation d’acide gras, qui deviendront dès la naissance les grands pourvoyeurs d’énergie myocardique.
L’endocarde tapisse les cavités auriculaires et ventriculaires ; il est constitué de cellules endothéliales qui sont de véritables glandes autocrines interagissant avec le muscle cardiaque et les myocytes, via les protéines membranaires. Grâce à ses propriétés antiagrégantes plaquettaires, l’endocarde empêche les thrombus intracardiaques.
Physiopathologie
Adaptation de la masse myocardique au travail du cœur. L’adaptation vise à mettre en harmonie la masse myocardique avec le travail du cœur, de façon à normaliser la contrainte.
Physiologiquement, l’adaptation est basée sur la loi de Laplace, qui indique que la contrainte pariétale (CP), ou encore wall stress, qui constitue le véritable travail du cœur est directement fonction de la pression développée dans la cavité (P), du diamètre de cette cavité (D) et inversement fonction de l’épaisseur de la paroi (E) : CP = P x D : E.
Ainsi, avec l’augmentation progressive du débit (c’est-à-dire du diamètre) et de la pression, l’épaisseur de la paroi augmentera proportionnellement et la masse myocardique évoluera de façon harmonieuse (par hyperplasie chez le fœtus et le nouveau-né, et par hypertrophie secondairement). On peut considérer comme adaptation physiologique l’hypertrophie myocardique observée dans l’hypertension artérielle (systémique pour le ventricule gauche et pulmonaire pour le ventricule droit) ou les sténoses des valves aortiques ou pulmonaires.
Deux circonstances d’ada-ptation pathologique :
• la cellule myocardique a un mauvais rendement, la masse myocardique va augmenter pour compenser la faiblesse qualitative de chaque gramme de tissu. Il s’agit des myocardiopathies hypertrophiques observées en cas d’altération de la contractilité de la fibre myocardique, expliquant que l’on peut avoir une contractilité globale maintenue, voire augmentée, alors que la fibre elle-même est hypocontractile. C’est ce qui s’observe couramment dans les myocardiopathies hypertrophiques de l’adulte.
• la désadaptation du processus de normalisation de la contrainte pariétale avec :
- myocardiopathie hypertrophique si la normalisation se fait pour une contrainte basse,
- myocardiopathie hypotrophique (dilatée à paroi mince) lorsque que l’adaptation se fait pour une contrainte élevée. C’est ce qui permet d’expliquer que des sujets normaux peuvent avoir des fractions d’éjection relativement basses avec une masse myocardique faible alors que d’autres ont des fractions d’éjection élevées avec des masses myocardiques élevées.
Il est intéressant de noter que les études sur les fibres myocardiques prélevées après transplantation et étudiées individuellement in vitro sur chambre d’organe (étirement vs tension) ont une contractilité normale dans les myocardiopathies hypokinétiques à parois minces, alors que des fibres myocardiques sont hypocontractiles dans les myocardiopathies hypertrophiques et hyperkinétiques ce qui confirme bien que l’hypertrophie est plus un processus d’adaptation que le processus primitif de la CMH.
Définition physiologique et échographique des MCP
Une myocardiopathie peut provenir d’une altération des propriétés systoliques et diastoliques de la fibre myocardique ou d’une inadéquation de la masse myocardique par rapport à son volume et à son travail.
Théoriquement, on parle de MCPH lorsque la masse myocardique est augmentée. En pratique, on parle de MCPH lorsque les parois sont trop épaisses. Or, la paroi est très épaisse si la fonction systolique globale est préservée car la masse se répartit sur un volume normal. La paroi est moins épaisse si la fonction systolique est altérée car elle se répartit sur un volume plus grand et l’on assiste à une diminution apparente de l’hypertrophie de la paroi (épaisseur mais non masse) lorsque la fonction systolique s’altère. De nombreuses MCH de l’enfant évoluent vers des MC dilatées de l’adulte à paroi moins épaisses.
Myocardiopathie secondaire
Lorsque la myocardiopathie est dite secondaire, cette altération de la fonction ou de la masse est due à des facteurs extérieurs au muscle cardiaque, c’est-à-dire que l’origine de la MCPH provient :
• soit des conditions de charge (surcharge barométrique ou volumétrique due à une pathologie vasculaire ou à une anomalie des valves ou des cloisons du cœur, comme dans l’hypertension artérielle systémique ou pulmonaire et aux malformations congénitales ou acquises),
• soit d’une altération du rythme cardiaque — trop lent, trop rapide, trop irrégulier — de façon chronique ou responsable d’une désynchronisation de la contraction.
Myocardiopathie primitive acquise
Elle survient lorsque la fibre est atteinte :
• en raison d’une mauvaise irrigation de sang : c’est l’insuffisance coronarienne due à une anomalie des artères coronaires, congénitale ou acquise (myocardiopathie ischémique),
• ou altérée par un toxique (comme dans l’intoxication aux antracyclines),
• ou infectée (en général par un mécanisme immunologique) dans les myocardites.
Ces MCP sont en général peu hypertrophiques.
Myocardiopathie primitive constitutionnelle
Les fibres mêmes sont altérées :
• soit dans leur propriété (protéine contractile),
• soit dans leur architecture (cytosquelette),
• soit dans leur métabolisme (en particulier oxydatif par une anomalie de la mitochondrie ou du métabolisme des acides gras et leur transport).
Sur le plan théorique et scientifique cet article ne devrait traiter que les myocardiopathies primitives constitutionnelles, mais sur le plan pratique, il est crucial d’éliminer les myocardiopathies secondaires, qui sont plus souvent curables que les myocardiopathies primitives. Ils ont donc un impératif de les exclure dans la démarche vis-à-vis du patient.
Conduite à tenir devant les MCPH de l’enfant en fonction de l’âge
Chez le fœtus
L’échocardiographie peut les découvrir, en particulier lors de l’examen de la 22e ou de la 32e semaine.
La MCPH peut être « secondaire », en rapport avec une altération de la circulation du fœtus comme chez les jumeaux transfuseur-transfusé ; elle peut aussi être la conséquence d’une anomalie importante du placenta, ce qui augmente les résistances vasculaires du fœtus et la postcharge. Ces MCP guériront avec l’accouchement.
La MCPH peut être le fait d’une myocardiopathie constitutionnelle à expression très précoce, en particulier celles qui touchent le métabolisme glucidique du myocarde ; les myocardiopathies hypertrophiques de nouveau-nés de mère diabétique, ou la maladie de Pompe (glycogénose de type II) (figure 3).
Figure 3. Maladie de Pompe.
Il faut surtout souligner que la plupart des MCPH, en particulier les MCPH par anomalie des protéines contractiles, ne se déclarent pas dès la période fœtale et qu’en aucun cas, la constatation d’un VG normal chez un fœtus ne doit rassurer en cas d’antécédents familiaux de MCPH.
À l’inverse, il faut savoir qu’à l’image de la MCPH du nouveau-né de mère diabétique, dans un tiers des cas environ, une MCPH fœtale chez une mère non-diabétique disparaîtra après la naissance avec un réel espoir de cœur normal.
Ces constatations ne facilitent ni le conseil, ni l’attitude à adopter en cas de diagnostic prénatal et l’option d’une IMG.
Chez le nouveau-né
Les MCPH sont peu fréquentes.
Il faut rechercher :
- le diabète maternel, avant tout, dont la MCPH très sévère guérira toute seule en quelques semaines ;
- les hypertrophies secondaires aux HTA en particulier de la coarctation de l’aorte ou de la sténose ou thrombose de l’artère rénale ;
- la maladie de Pompe, bien que très rare, (figure 3) qui peut bénéficier d’un traitement enzymatique efficace et sera fortement suspectée à cet âge sur un ECG très volté (figure 4) et confirmé par le dosage de la maltase acide.
Figure 4. Grands voltages des QRS (maladie de Pompe).
- Les MCP métaboliques ne sont pas fréquentes à cet âge en dehors d’un contexte franc de maladie métabolique avec hypoglycémie, hyperlactacidémie. Les MCP sont en règle hypokinétiques à parois minces ou peu épaisses. Les maladies des acides gras (métabolique ou transfert par la carnitine) ne s’expriment qu’après quelques semaines.
En cas de signe de défaillance cardiaque ou de collapsus, le nouveau-né doit être transféré dans un service spécialisé ; des prélèvements de sang et d’urine ainsi qu’une biopsie cutanée pour culture de fibroblaste doivent être conservés, et en cas de décès, des biopsies de muscle, de foie et si possible de myocarde doivent être pratiquées et conservées dans le formol et l’azote liquide pour histologie et biochimie (en particulier mitochondriale). C’est souvent le seul moyen de faire un diagnostic permettant un conseil génétique.
Chez le nourrisson
Les MCPH très hypertrophiques et hyperkinétiques sont rares. On retrouve les principales causes du nouveau-né, en particulier les MCPH du diabète maternel, qui sont en voie de régression, et celles de la maladie de Pompe, dont la séméiologie s’enrichit de symptômes neurologiques et musculaires qui peuvent être l’occasion du diagnostic (hypotonie et macroglossie) avec, à ce stade, un pronostic très péjoratif.
À cet âge, la découverte d’une MCPH très hypertrophique, hyperkinétique et éventuellement obstructive doit faire rechercher 3 étiologies principales :
- une MCPH secondaire à une augmentation de la postcharge, en particulier une HTA (coarctation ou néphropathie) ou un obstacle aortique ;
- une MCPH familiale à expression précoce (la plupart ne deviennent hypertrophiques que chez le grand enfant), c’est le caractère asymétrique et surtout l’enquête familiale qui permettent le diagnostic ;
- un syndrome de Noonan, dont la séméiologie et l’échographie ressemblent beaucoup aux MCPH de l’adulte avec en plus la dysmorphie (figure 5) et quelques particularités cardiaques (une sténose valvulaire pulmonaire et un axe de QRS en AVR). Il est probable que le mécanisme de la MCPH du syndrome de Noonan fasse intervenir une anomalie de protéines contractiles car elle mime la MCPH classique et pose les mêmes problèmes de risque de mort subite, de surveillance et de traitement. Dans notre expérience, cette étiologie est plus fréquente chez le nourrisson que la MCPH classique, en particulier dans les formes qui nécessitent une levée de l’obstruction sous-aortique.
Figure 5. Syndrome de Noonan.
MCPH moins hypertrophiques et hypokinétiques.
• En l’absence de ces 5 étiologies (diabète maternel, maladie de Pompe, syndrome de Noonan et forme familiale de MCPH et MCPH secondaire à une HTA), les MCPH du nourrisson sont en règle moins hypertrophiques car souvent hypokinétiques (la masse se répartissant sur un plus large volume) et imposent une hospitalisation en service spécialisé à la recherche d’une anomalie métabolique et en particulier mitochondriale.
Le diagnostic repose sur :
• la recherche d’une atteinte non cardiologique, en particulier neurologique (IRM), musculaire, oculaire, hépatique, hématologique,
• la recherche d’une anomalie métabolique (oxydoréduction avec acidose lactique avec rapports anormaux lactate/piruvate et acéto-acétate/glutarate),
• sur une enquête familiale car ces maladies sont génétiques (malheureusement le plus souvent récessives autosomiques donc sans expression chez les parents).
• En l’absence de ces anomalies métaboliques et extracardiologiques, on ne peut éliminer une MCPH mitochondriale à expression cardiaque pure ou très prédominante. Il faut alors avoir recours à la biopsie du myocarde pour faire le diagnostic en étudiant le métabolisme mitochondrial avec sa cascade caractéristique (figure 6), jugé sur les rapports d’oxydoréduction des différents complexes (1, 2, 3 et 4), et ce sur les fragments de muscle vivant saisis à la biopsie et congelés dans l’azote liquide. Dans notre expérience, cette étiologie est présente dans près des deux tiers des MCPH hypokinétiques.
• Parmi ces MCP mitochondriales, il est important de souligner le cas du syndrome de Barth, qui est une forme récessive liée au sexe avec neutropénie.
• Les autres MCP observées ne sont en règle pas très hypertophiques ou d’aspect très particulier :
- les anomalies métaboliques non mitochondriales (acide gras) se traduisent en général par des MCP hypokinétiques peu ou pas hypertrophiques, voire par des troubles du rythme isolés. Leur diagnostic repose sur le contexte métabolique (acidose avec hypoglycémie hypocétosique) et sur la chromatographie des acides organiques urinaires, sur le profil acylcarnitine et l’étude de la bêta-oxydation des lymphocytes et des fibroblastes. Parmi ces anomalies du métabolisme des acides gras, il faut insister sur les myocardiopathies par déficit en carnitine, qui ne sont en général pas hypertrophiques mais qui guérissent par apport en L-carnitine ;
- les MCP par persistance de myocarde spongieux (ou non compacté) peuvent prendre la forme de MCPH si l’on ne remarque pas l’aspect très particulier du myocarde très trabéculé et proliférant (figure 2).
Figure 6. Oxydo-réduction au niveau de la mitochondrie.
Chez l’enfant
Diabète et maladie de Pompe ont disparu, persistent le syndrome de Noonan, les MCP mitochondriales, avec en particulier la maladie de Friedreich, et se confirment les MCPH de type adulte par anomalies des protéines contractile du myocarde.
La maladie de Friedreich (hérédodégénérescence spinocérébelleuse) comporte presque constamment une MCPH qui peut être inaugurale (les symptômes neurologiques pouvant rester discrets longtemps avec un simple et léger trouble de l’équilibre).
Le diagnostic se fait sur la clinique neurologique (abolition des réflexes ostéotendineux, déséquilibre), constatation des pieds creux. Une fois évoqué, le diagnostic est confirmé par l’identification de la délétion génique sur le gène de la frataxine sur le chromosome 9enq13-q21).
Le mécanisme de la MCPH est maintenant connu. Il s’agit d’une altération du métabolisme fer-soufre avec accumulation du fer dans la mitochondrie et un appauvrissement du fer dans le cytosol. L’accumulation de fer et de ses radicaux libres altère le fonctionnement de la mitochondrie d’où une baisse énergétique de la fibre compensée par l’hypertrophie myocardique. Faire ce diagnostic est important car le pronostic (moteur, équilibre, scoliose) est très péjoratif contre-indiquant une transplantation. En outre, il s’agit d’une maladie récessive autosomique qui se prête à un diagnostic prénatal (conseil génétique). Enfin, la MCPH peut être améliorée et du moins stabilisée par un traitement anti-oxydant non réducteur (Mnesis® ou idébénome).
Les MCP avec fibrose endocardique observées dans les hyperéosinophilies qui peuvent être tapissées d’un matelas de thrombose épaississant le myocarde avec un profil restrictif (figure 7).
Figure 7. Fibrose myocardique avec profil restrictif du Doppler.
La MCPH « classique » : en pratique, il y a 2 situations :
• la famille est connue pour avoir une MCPH et on veut savoir si l’enfant est atteint : en dehors des cas où l’analyse génétique est possible voire le gène déjà identifié dans la famille, c’est le phénotype cardiaque qui permettra de répondre en révélant la présence d’anomalie de l’ECG ou de l’échographie, tout en sachant que lorsque ces anomalies ne sont pas présentes, il est impossible d’innocenter le patient avant qu’il ait atteint l’âge adulte ;
• la MCPH est découverte chez un enfant non dysmorphique, sans anomalie apparente extra-cardiaque et la question est de savoir si cette MCPH est autosomique dominante par anomalie d’un gène sur les protéines contractiles : la découverte d’une anomalie même asymptomatique chez un des parents est déterminante. En son absence, il est impossible d’éliminer le diagnostic car l’enfant peut être le premier à avoir la mutation ou celle-ci n’est pas exprimée chez l’un des parents qui a l’anomalie génétique.
Aujourd’hui, on peut tester ces hypothèses en confiant le sang de l’enfant et des parents au centre de la Pitié-Salpétrière pour tester une batterie des gènes les plus fréquents et ainsi avoir une réponse génétique dans plus de la moitié des cas. Toutefois, si l’enquête génétique est négative, on ne peut exclure le diagnostic car tous les gènes ne sont pas connus et tous les gènes connus ne sont pas testés.
En pratique
Une fois la MCPH diagnostiquée ou très probable (forme familiale ou enquête négative) se posent les questions fondamentales concernant le pronostic, la surveillance, les autorisations d’activités physiques et sportives, et les éventuels traitements.
Le pronostic est incertain en raison du risque de mort subite qui est globalement plus élevé chez l’enfant que chez l’adulte et s’établit autour de 6 %/an. Les éléments défavorables sont les antécédents de mort subite avortés chez l’enfant ou révélés dans la famille, une mauvaise adaptation à l’effort, une hypertrophie considérable avec une obstruction sous-aortique, des ponts coronaires ou encore une anomalie génétique particulièrement sévère.
La surveillance découle de ces facteurs aggravants du pronostic. Un vrai bilan de départ comprend, outre l’échographie, une épreuve d’effort, un Holter et éventuellement une coronarographie. Ensuite la surveillance doit être annuelle avec :
- une échographie-Doppler pour déterminer le degré d’hypertrophie et d’obstacle sous-aortique, l’apparition d’une IM ou de signes de dysfonction systolique ou diastolique,
- une épreuve d’effort à la recherche de troubles du rythme, et surtout d’une mauvaise adaptation de la pression artérielle à l’effort, et de malaise à la récupération.
Les autorisations dépendent en particulier de l’épreuve d’effort mais, même si celle-ci est négative, le principe de précaution (qui mine notre société) impose une grande prudence, et l’interdiction des sports de compétition et l’inscription dans des clubs sportifs. En revanche, en l’absence de signes de mauvais pronostic, on peut autoriser l’activité physique à l’école avec autolimitation pour une bonne socialisation et un bon épanouissement de l’enfant.
Les traitements sur le risque de mort subite sont symptomatiques et leur efficacité n’est pas démontrée (ni plus que leur inefficacité car aucune étude randomisée n’a été publiée).
En cas d’obstruction, les bêtabloquants sont essayés et, en cas d’échec, nous avons tendance à préconiser une levée de l’obstacle.
Nous n’avons pas d’expérience en pédiatrie du pacing pour remodeler le ventricule. Nous optons, en général, pour la chirurgie qui peut être pratiquée à tout âge, mais il faut des équipes très entraînées pour une bonne résection sans BAV en chirurgie (septectomie antérieure chirurgicale). Une autre option chez le grand enfant est la nécrose du septum par embolisation des premières septales (alcoolisation ou microsphère) avec une résection bien calibrée (l’écho de contraste dans la septale avant embolisation permet de prédire le territoire nécrosé).
En cas de troubles du rythme « légers », certaines équipes recommandent la cordarone.
En cas de troubles du rythme graves documentés, ou en cas de syncopes graves enregistrées, ou dans certaines familles à risque élevé (antécédents, génétiques) se discute un défibrillateur que l’on peut envisager chez l’enfant à partir de 15 kg.
Enfin, dans quelques cas, la transplantation cardiaque peut se discuter, en particulier en cas de défaillance cardiaque par dysfonction systolique ou diastolique.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
