




Publié le 28 fév 2013Lecture 11 min
Les cardiomyopathies restrictives : de quoi s’agit-il ?
D. MOHTY, CHU Dupuytren, Limoges
JESFC
Les cardiomyopathies restrictives (CMR) font partie d’un ensemble de maladies rares et diverses du myocarde, dont le point commun est une rigidité pariétale empêchant le myocarde de se relaxer de façon effective.
La conséquence hémodynamique de cette rigidité est une augmentation importante et rapide des pressions de remplissage du ventricule gauche (VG) sans augmentation suffisante, voire avec diminution des volumes ventriculaires. Initialement, la fonction systolique du VG ou plutôt la fraction d’éjection du VG (FEVG) est préservée, contrastant avec une altération précoce de la fonction diastolique. Les parois ventriculaires peuvent être épaissies ou normales selon le processus et la cause de la cardiopathie.
Le ventricule droit (VD) peut être également atteint avec souvent une prédominance des signes droits. Le diagnostic doit être systématiquement évoqué en présence d’un tableau d’insuffisance cardiaque globale en l’absence de cardiomégalie et de baisse de la FEVG.
Même si le traitement des cardiopathies restrictives reste peu satisfaisant, il est fondamental de faire le diagnostic différentiel avec la péricardite constrictive (PCC) avec laquelle existent des similarités cliniques et hémodynamiques, notamment la physiologie restrictive, mais dont la thérapeutique est complètement différente.
La cardiomyopathie restrictive reste la moins fréquente parmi les autres types de cardiomyopathies : dilatées, hypertrophiques(1). En dehors des pays tropicaux, l’amylose cardiaque systémique reste la cause la plus fréquente de CMR. La cardiomyopathie restrictive idiopathique est la plus rare et la plus difficile à diagnostiquer. La fibrose endomyocardique est une cardiopathie endémique dans certaines régions : Afrique, Inde, Amérique du Sud et centrale. Dans la sarcoïdose et l’hémochromatose, ce sont les signes extracardiaques qui font suspecter souvent l’étiologie de la CMR.
L’échocardiographie est l’examen clé pour une description méticuleuse des anomalies typiques de la cardiomyopathie restrictive. L’IRM cardiaque est réalisée en complément de l’échographie en raison de son excellente caractérisation tissulaire. Elle permet de mettre en évidence le rehaussement tardif dû à la fibrose myocardique commune à toutes les étiologies mais dont la localisation permet d’orienter vers le diagnostic étiologique des CMR.
Classification
Les CMR peuvent résulter de différentes pathologies systémiques rares, dont la plupart ne seront jamais ou très rarement rencontrées en pratique clinique. Certaines étiologies sont plus fréquentes, notamment l’amylose à chaînes légères, dont la principale complication conditionnant le pronostic est une CMR avec insuffisance cardiaque résultant de dépôts anormaux de fibrilles amyloïdes au niveau interstitiel à l’origine de la rigidité pariétale. À l’inverse, dans la CMR idiopathique, les altérations hémodynamiques peuvent survenir en l’absence de modifications histologiques évidentes. De nombreuses autres maladies donnent un tableau de cardiopathies avec physiologie restrictive. Celles-ci peuvent être classées en fonction de l’atteinte myocardique versus endomyocardique ou de l’atteinte infiltrative versus non infiltrative ou maladie de surcharge.
Le tableau 1 adapté de S.S. Kushwaha et coll.(2) résume les principales étiologies des cardiopathies restrictives.
La CMR idiopathique(3) est très rare et difficile à affirmer ; elle donne une physiologie restrictive sévère sans modification histologique spécifique, ni infiltration, ni surcharge myocardique secondaire identifiable sur la biopsie myocardique, qui retrouve une fibrose interstitielle non spécifique, associée dans certains cas à une hypertrophie modérée des myocytes qui peut faire penser à tort à une cardiopathie hypertrophique familiale sarcomérique. L’ETT ne retrouve pas d’hypertrophie des parois ventriculaires, mais un VG de taille et de contractilité globalement préservées contrastant avec une dilatation atriale importante, un profil mitral restrictif et une hypertension artérielle pulmonaire systolique. La survie des patients avec CMR idiopathique est réduite à 64 % à 5 ans d’après le travail de la Mayo Clinic et significativement corrélée au sexe masculin, à une classe fonctionnelle NYHA III ou IV, à une OG > 60 mm et à une HTAPs(3). A contrario, la cardiopathie amyloïde (surtout AL à chaînes légères) est la cause de cardiomyopathie restrictive la plus fréquente en Occident(4).
Par ailleurs, les cardiopathies hypertensives, dilatées, ou ischémiques sont beaucoup plus fréquentes, et peuvent aussi donner à un stade avancé une physiologie restrictive pouvant mimer celle des cardiomyopathies restrictives.
Diagnostic clinique
Les patients atteints d’une CMR présentent en plus de certains symptômes propres à chaque étiologie, souvent une asthénie importante, une dyspnée paroxystique ou nocturne, voire une orthopnée ou des œdèmes des membres inférieurs. Généralement, les patients n’ont pas de douleur thoracique, sauf ceux ayant une amylose car celle-ci peut occasionner une atteinte de la microcirculation coronaire, alors que les gros troncs coronaires sont macroscopiquement sains. À un stade avancé, tous les signes d’insuffisance cardiaque peuvent être présents sauf la cardiomégalie.
ECG
Les anomalies électriques des cardiopathies restrictives sont souvent variées selon le processus étiologique. Il peut s’agir d’anomalies de la repolarisation du segment ST ou des ondes T peu spécifiques ou de troubles de la conduction.
La fibrillation atriale paroxystique ou persistante n’est pas rare (> 70 % des cas) et elle est due au remodelage atrial. Mais quand le patient est en rythme sinusal, des ondes P amples peuvent être visibles.
Par ailleurs, certaines anomalies peuvent orienter vers tel ou tel diagnostic ; par exemple, un microvoltage avec un aspect d’ondes R rabotées ou pseudoinfarctus orienterait vers une amylose cardiaque, alors qu’un PR court avec aspect « d’HVG électrique » orienterait vers une maladie de Fabry.
Radiographie du thorax
Elle n’aide pas pour le diagnostic étiologique mais elle confirme l’absence de cardiomégalie, sauf à un stade avancé où la dilatation auriculaire est bien marquée. On peut voir un syndrome interstitiel en rapport avec une congestion veineuse ou interstitielle. Les calcifications péricardiques sont importantes à rechercher en cas de diagnostic différentiel de péricardite constrictive (PCC).
Marqueurs biologiques
Le dosage du BNP ou NTproBNP peut aider au diagnostic en confirmant l’origine cardiogénique de la dyspnée. En outre, il aide surtout dans le diagnostic différentiel puisqu’il est augmenté dans la CMR, mais normal en cas de PCC, malgré une présentation clinique et hémodynamique similaire. Une insuffisance rénale inexpliquée permet par exemple d’orienter vers une maladie de Fabry. D’autres perturbations biologiques sont à rechercher en cas de suspicion étiologique particulière (ferritine plasmatique, électrophorèse et immunoélectrophorèse sérique, dosage de l’alpha galactosidase, etc.).
Échocardiographie
Les signes échographiques à rechercher en cas de suspicion de cardiomyopathie restrictive incluent :
- des ventricules généralement de taille normale ;
- une FEVG souvent préservée ou discrètement altérée ;
- des parois ventriculaires épaissies ou non en fonction du processus étiologique ;
- une dilatation bi-auriculaire marquée, par augmentation des pressions de remplissage du VG et du VD ;
- une dysfonction atriale gauche, présente dans l’amy lose cardiaque primitive, due à une véritable myopathie atriale causée par le dépôt des chaînes légères toxiques ;
- un profil mitral typiquement restrictif avec onde E de remplissage rapide augmentée et onde A diminuée, E/A > 2, temps de décélération raccourci < 150 m/s ;
- un effondrement des vitesses myocardiques au niveau de l’anneau mitral avec e’ souvent < 3 cm/s, une onde S < 5 cm/s ;
- un rapport E/E’ (septal et/ou latéral) augmenté > 15 ;
- une diminution de la composante systolique et une augmentation de la composante diastolique du flux des veines pulmonaires comme au niveau du flux des veines sus-hépatiques ;
- le 2D « strain », ou déformation longitudinale globale du VG, est souvent altéré, et s’est avéré un facteur pronostique de survie majeur et indépendant dans la cardiopathie amyloïde(5). De plus, les anomalies régionales du strain peuvent orienter vers tel ou tel diagnostic : l’amylose aboutit typiquement à une baisse du strain longitudinal des segments basaux avec préservation relative du strain apical(6), contrairement à l’atteinte due à la maladie de Fabry qui donnerait volontiers une altération plus localisée de la déformation de la paroi inféro-latérale ;
- un péricarde a priori normal non épaissi.
L’IRM cardiaque joue un rôle essentiel
L’IRM cardiaque permet le diagnostic étiologique des cardiomyopathies restrictives en raison de sa meilleure caractérisation tissulaire, excellente résolution temporale et spatiale, et le diagnostic différentiel avec la PCC dont les conséquences hémodynamiques peuvent être similaires. En effet, l’ETT est souvent prise en défaut pour visualiser un épaississement péricardique, contrairement à l’IRM ou au scanner. D’ailleurs, il est recommandé de réaliser une IRM cardiaque pour le diagnostic différentiel entre PPC et CMR. De plus, les autres signes de la CMR peuvent être visualisés en IRM telle la dilatation bi-auriculaire, la taille des ventricules, l’épaississement des parois et la dilatation du système cave, avec une meilleure résolution temporospatiale.
Le cathétérisme cardiaque
La réalisation du cathétérisme n’est plus obligatoire pour confirmer le diagnostic. Quand il est réalisé, il montre une augmentation des pressions de remplissage du VG (> 12 mmHg) et du VD (> 15 mmHg) généralement. L’aspect de dip-plateau, typique de la CMR quelle que soit son étiologie, mais aussi de la PCC, est caractérisé par une diminution précoce, rapide et profonde des pressions ventriculaires en tout début de diastole suivie d’une augmentation rapide des pressions jusqu’à un plateau qui atteint au moins un tiers du pic de la pression systolique ventriculaire droite. Cet aspect peut être également visible sur les courbes auriculaires. La pression auriculaire gauche dépasse habituellement 15 mmHg, la pression atriale droite > 7 mmHg et la pression capillaire pulmonaire > 12 mmHg.
Contrairement à la PCC, dans la CMR, il n’y a pas d’égalisation des pressions diastoliques des cavités droites et gauches ni des variations respiratoires des pressions.
Diagnostic différentiel
Il faut toujours garder à l’esprit que la PCC peut se présenter au niveau clinique comme la CMR. En effet, la CMR et la PCC sont caractérisées par un profil clinique et hémodynamique commun d’adiastolie, entraînant souvent des difficultés diagnostiques. Cliniquement, la symptomatologie fonctionnelle est aspécifique (dyspnée, asthénie, hépatalgies), de même que l’examen physique qui montre un tableau d’insuffisance cardiaque globale à prédominance droite. Sur le plan des paramètres paracliniques, le tableau 2 résume les principales différences entre CMR et PPC, notamment au niveau électrocardiographique, échocardiographique(7), IRM/TDM et cathétérisme cardiaque. Il est important d’utiliser tous les paramètres afin de faire le diagnostic différentiel entre la CMR et la PCC.
Cependant, il est encore des situations cliniques difficiles, par exemple de cardiomyopathie post-radique, où il y a une intrication, voire coexistence des deux physiologies de CMR et de PCC, et dans toutes les situations qui rendent difficiles l’analyse des variations respiratoires des flux Doppler (FA rapide, BPCO, obésité sévère).
Pronostic
Le pronostic des CMR, dépend de l’étiologie à l’origine de la CMR, mais il est généralement sombre en présence de signes d’insuffisance cardiaque avec une mortalité précoce importante(8).
Traitement symptomatique
Les diurétiques sont les seuls utilisables pour améliorer la symptomatologie des patients, malgré le risque possible d’hypotension.
Les IEC et les bêtabloquants sont à utiliser avec précaution surtout dans la cardiopathie amyloïde où le risque d’hypotension est important(8). Les digitaliques ne sont indiqués qu’en cas de fibrillation auriculaire et sont contre-indiqués en cas d’amylose, en raison de la sensibilité particulière de ces patients à ces molécules. L’amiodarone peut être utilisée en cas de troubles de rythme supraventriculaires ou ventriculaires persistants. Le traitement anticoagulant est indiqué en cas de thromboses intracavitaires ou d’embolies artérielles ou pulmonaires documentées et d’arythmie atriale documentée.
Le stimulateur cardiaque définitif est indiqué en cas de troubles conductifs sévères.
Le traitement spécifique dépend de l’étiologie sous-jacente.
La transplantation cardiaque est à discuter si la cardiopathie et l’insuffisance cardiaque sont au stade terminal, au cas par cas selon la gravité et l’étiologie de la maladie, les comorbidités du patient et son espérance de vie.
En pratique
Les cardiomyopathies restrictives sont beaucoup moins fréquentes que les autres cardiomyopathies.
Leur diagnostic n’est pas toujours évident, surtout quand la restriction myocardique et la constriction péricardique sont présentes chez le même patient comme dans la cardiopathie post-radique ; d’où l’intérêt d’une évaluation globale, minutieuse et multiparamétrique, qui permettra d’avoir une orientation étiologique.
L’apport des nouvelles techniques échocardiographiques ainsi que celui de l’IRM cardiaque avec injection de gadolinium ont permis d’améliorer la précision diagnostique et étiologique de ces cardiomyopathies.
La conduite à tenir dépend de l’étiologie pour les CMR, mais globalement, le plus important reste de les différencier des PCC dont le traitement curatif est une péricardectomie chirurgicale.
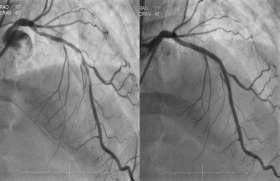
Figure 1. Exemple d’une cardiomyopathie restrictive, infiltrative, sévère, due à une infiltration amyloïde de chaînes légères AL.
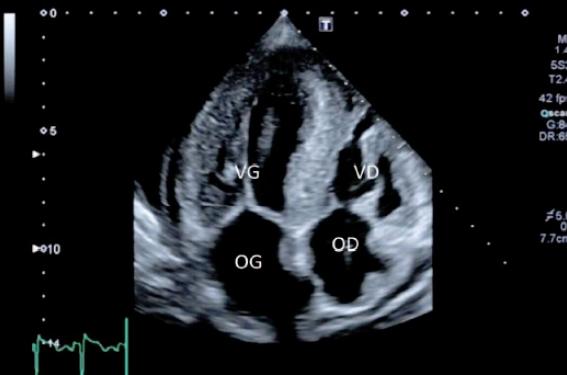
Figure 2. Vue petit axe et parasternale grand axe respectivement, de la même patiente, montrant une cavité ventriculaire réduite, l’épaississement des parois ventriculaires, le petit épanchement péricardique, la dilatation atriale et l’aspect « hyperéchogène » des parois du VG.
Figure 3. A. Profil normal pseudonormalisé, type II, TD raccourci. B. Effondrement des vitesses myocardiques par le doppler tissulaire à l’anneau mitral latéral.
Figure 4. Acquisition des 4, 3 et 2 cavités en 2D speckle tracking du VG d’un patient atteint d’une cardiopathie amyloïde AL avancée ; le 2D strain global longitudinal est abaissé à -10 % alors que l’apex paraît relativement préservé (vue en oeil-de-boeuf des différents segments du VG avec un déformation à l’apex codé en rouge = préservée comparé aux segments basaux codés en rose = altéré).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
