

Publié le 15 oct 2020Lecture 13 min
Recommandations sur la prise en charge des cardiopathies congénitales de l’adulte - Ce que le cardiologue doit savoir
Reaksmei LY, Laurence ISERIN, département de cardiologie, HEGP, Paris
La population des adultes avec cardiopathies congénitales ne cesse de croître grâce aux progrès des avancées médicales, technologiques et chirurgicales (figure 1), avec une prévalence estimée de l’ordre de 3 000 par million d’habitants, dépassant celle de la population pédiatrique. Plus de 90 % des enfants naissant avec une cardiopathie congénitale survivent en effet à l’âge adulte, mais ils ne sont pas tous guéris.
Figure 1. Chronologie des chirurgies des cardiopathies congénitales.
Le suivi de cette population reste donc indispensable et nécessite une évaluation tout du moins pour les cardiopathies modérées et complexes (figure 1) par un spécialiste des cardiopathies congénitales de l’adulte, du fait de l’évolutivité intrinsèque de ces cardiopathies et des séquelles hémodynamiques et/ou rythmiques liées à la chirurgie. Tout au long de sa vie, le patient porteur d’une cardiopathie congénitale est confronté à des problématiques multiples et variées : la nécessité d’un nouveau geste chirurgical ou de cathétérisme interventionnel pour des lésions résiduelles, les déroulements de la grossesse et ses risques, pour la mère et le foetus, diagnostic anténatal pour la descendance, les maladies cardio-vasculaires acquises associées, le risque d’endocardite infectieuse, l’insuffisance cardiaque, l’arythmie, et enfin la qualité de vie et ses répercussions psychologiques et sociales.
Les nouvelles recommandations de l’European Society of Cardiology sur le sujet, parues en août 2020, préconisent que tout patient avec une cardiopathie congénitale soit évalué au moins une fois dans un centre expert multidisciplinaire spécialisé en cardiopathie congénitale de l’adulte. Celui-ci doit comprendre des cardiologues spécialisés aguerris aux cardiopathies congénitales, des radiologues, cathétériseurs interventionnels, rythmologues, chirurgiens cardiaques… afin de déterminer le niveau de prise en charge. Ce dernier se divise en trois niveaux, se basant principalement sur la complexité de la cardiopathie présentée en tableau 1 : (i) patient qui doit être suivi dans un centre expert ; (ii) patient dont le suivi peut être partagé entre un centre expert et un centre de cardiologie générale ; (iii) patient pouvant être suivi par un cardiologue généraliste.
L’évaluation initiale par le cardiologue
Savoir ce qui s’est passé dans l’enfance est un élément crucial pour comprendre la situation clinique actuelle du patient adulte avec une cardiopathie congénitale, en récupérant notamment ses comptes rendus médicaux et chirurgicaux.
L’examen physique doit comprendre la mesure de la saturation en oxygène percutanée pour distinguer une cardiopathie cyanogène ou d’une cardiopathie non cyanogène, la mesure de la pression artérielle (notamment au niveau du bras droit pour les coarctations aortiques), et doit être attentif aux signes d’insuffisance cardiaque, en particulier droite.
L’électrocardiogramme permet de dépister des troubles de conduction, ou de rythme, d’analyser la largeur des QRS qui est un facteur pronostique dans la tétralogie de Fallot par exemple (bloc de branche droit).
L’échographie cardiaque transthoracique reste l’examen d’imagerie de référence dans l’évaluation initiale, car elle donne des renseignements clés sur l’anatomie cardiaque, la présence de défects, de lésions résiduelles, ainsi que le fonctionnement des ventricules, des valves et des différents montages chirurgicaux. Mais chez l’adulte, cela doit souvent être complété par des imageries en coupe comme le scanner et l’IRM cardiaque qui doivent être réalisés par des radiologues accoutumés à ces patients.
Les cardiopathies congénitales les plus fréquemment rencontrées
Les shunts gauche-droit
Communication inter-atriale (CIA)
La communication inter-atriale est la cardiopathie congénitale la plus fréquente, si on s’affranchit de la bicuspide aortique, avec une prévalence estimée à environ 1 personne sur 25 000. Dans près de 80 % des cas, il s’agit d’une CIA ostium secundum, c’est-à-dire située au milieu du septum inter-atrial, dans la région du foramen oval. Il est important de rechercher d’autres anomalies associées, notamment un retour veineux pulmonaire normal, une sténose pulmonaire ou une anomalie de la valve mitrale.
Le shunt dans le sens gauchedroit est expliqué par une compliance naturelle meilleure du ventricule droit comparée au ventricule gauche, entraînant dans un premier temps une surcharge volumétrique/dilatation des cavités droites et plus tardivement au terme de décennies d’hyperdébit pulmonaire, une hypertension pulmonaire, qui doit être impérativement explorée, lorsqu’elle existe.
L’échographie permet d’identifier la taille de la CIA, sa localisation (figure 2), et le retentissement hémodynamique. Les critères de dilatation des cavités droites sont les mêmes qu’en cardiologie conventionnelle (surfaces, rapport VD/VG, diamètres du ventricule droit, et de l’anneau tricuspide...). L’imagerie en coupe (IRM cardiaque préférentiellement) permet d’évaluer les volumes du ventricule droit, ou de rechercher un retour veineux pulmonaire anormal en cas de doute.
Figure 2. Les différentes formes de CIA selon leurs localisations.
En cas de symptômes et d’une dilatation des cavités droites et en l’absence d’hypertension pulmonaire certaine, il est recommandé de fermer la CIA ostium secundum, en première intention par cathétérisme cardiaque.
Communication inter-ventriculaire (CIV)
La communication inter-ventriculaire est principalement diagnostiquée dans l’enfance avec une incidence de 5 pour 1 000 naissances. Elle se ferme souvent spontanément avant l’âge adulte. Il existe quatre types de CIV selon sa localisation (figure 3) :
– péri-membraneuse : la plus fréquente (soit 80 %), localisée dans le septum membraneux ;
– trabéculée : dans le septum interventriculaire musculaire ;
– de l’outlet : retrouvée dans les cardiopathies dites cono-troncales comme dans la tétralogie de Fallot ;
– de l’inlet : typique des CIV dans les canaux atrio-ventriculaires. Il est licite de rechercher d’autres anomalies associées, car les associations sont très fréquentes, telles qu’un obstacle de la voie droite.
Figure 3. Anatomie et classification des CIV. D’après Soto B et al. Br Heart J 1980 ; 43(2) : 332-43.
Le shunt d’une CIV va dans le sens gauche-droit car les résistances artérielles pulmonaires sont bien plus basses que les résistances artérielles systémiques. La conséquence est l’augmentation de la précharge dans les cavités gauches et donc leurs dilatations. L’échographie cardiaque permet de localiser la CIV, de rechercher des lésions associées, la présence d’une fuite aortique, et le retentissement hémodynamique, notamment la présence d’une hypertension pulmonaire.
Sur le plan thérapeutique, une fermeture de CIV est recommandée en cas de dilatation des cavités gauches, symptomatiques, d’une progression d’une fuite aortique secondaire à la CIV, d’un antécédent d’une endocardite infectieuse, en s’assurant toujours qu’il n’y ait pas d’hypertension pulmonaire. Dans ce cas, le patient doit être évalué dans un centre expert.
La fermeture est le plus souvent chirurgicale, mais les techniques percutanées existent notamment dans les CIV résiduelles.
Canal atrio-ventriculaire (CAV) et trisomie 21
Les canaux atrio-ventriculaires, en particulier complets (c’est-àdire CIV + CIA ostium primum) sont retrouvés chez les patients avec une trisomie 21.
L’association des deux shunts, à l’étage atrial et à l’étage ventriculaire, entraîne une surcharge à la fois des cavités droites et des cavités gauches, et un risque plus important de développer de l’hypertension pulmonaire. La présence d’un anneau atrio-ventriculaire unique est à l’origine d’une anomalie des feuillets valvulaires pouvant être responsable d’une fuite sévère.
En somme, les patients atteints d’un CAV opéré ou natif doivent être évalués dans un centre expert de cardiopathies congénitales de l’adulte.
Les obstacles gauches
Sténoses aortiques, sous-valvulaires et supra-valvulaires aortiques
L’évaluation de ces obstacles au niveau hémodynamique est sensiblement la même que pour l’évaluation d’une sténose aortique dégénérative avec les mêmes critères de sévérité et d’indications chirurgicales.
Ces sténoses amènent à rechercher les autres obstacles (coarctation, bicuspide aortique, anomalies mitrales…).
Notons que les sténoses supravalvulaires aortiques peuvent se manifester dans le cadre d’anomalies de l’élastine, comme dans le syndrome de Williams- Beuren. Ces patients doivent donc être orientés dans un centre de référence.
Coarctation aortique
La coarctation aortique est définie par un rétrécissement au niveau de l’isthme aortique. Elle doit être considérée comme une artériopathie généralisée et peut être associée aux autres obstacles gauches du coeur (associée jusqu’à 85 % de bicuspide aortique par exemple).
Le diagnostic est porté par la mesure du gradient tensionnel clinique entre les pressions artérielles des membres supérieurs (surtout du bras droit, car l’artère sous-clavière ipsilatérale a souvent été sacrifiée en cas de chirurgie antérieure) et les pressions artérielles des membres inférieurs. Il est considéré comme significatif s’il est supérieur à 20mmHg. Les pouls fémoraux sont souvent plus faibles qu’aux membres supérieurs. Le gradient échographique n’a que peu de valeur. C’est la présence d’un prolongement diastolique qui évoque une sténose isthmique significative (il n’existe parfois pas chez l’adulte en raison du développement d’une collatéralité thoracique parfois importante). Dans ce cas, il est quasi-indispensable de demander une imagerie en coupe (IRMou TDMaortique) pour analyser l’anatomie de l’aorte, la présence de collatéralités ou de complications associées (figure 4).
Figure 4. Coarctation aortique native serrée. L’intérêt de l’imagerie multimodale. Échographie cardiaque, à gauche : sténose serrée isthmique avec prolongement diastolique ; TDM cardiaque 3D, à droite : montrant la sténose avec collatéralités.
En cas d’hypertension artérielle avérée (notamment par des mesures ambulatoires de la pression artérielle) chez des patients avec une coarctation aortique opérée ou native, il est nécessaire d’adresser ces patients dans un centre expert pour réaliser un cathétérisme cardiaque en vue d’une éventuelle prise en charge thérapeutique par stenting/dilatation.
Les indications de chirurgie de coarctation aortique par thoracotomie latérale sont plus rares chez l’adulte, à l’opposé de l’enfant.
Les obstacles droits
Sténoses pulmonaires
La sténose valvulaire pulmonaire est souvent une lésion isolée, pouvant éventuellement être associée à des syndromes génétiques comme le syndrome de Noonan. Très souvent, l’échographie cardiaque est suffisante pour le diagnostic.
Lorsque la sténose est sévère (soit plus de 64 mmHg de gradient maximal) et symptomatique, la valvuloplastie percutanée au ballon est la technique thérapeutique de choix, réalisée dans un centre expert.
Quel que soit le niveau de la sténose pulmonaire (sous-valvulaire, valvulaire, supra-valvulaire ou des branches pulmonaires), il est important d’être attentif aux symptômes, à la fonction du ventricule droit ou à l’évolution d’une fuite tricuspide, car elle peut amener à un traitement interventionnel ou chirurgical. Mais dans tous les cas, elle doit également être évaluée dans un centre expert spécialisé dans les cardiopathies congénitales de l’adulte.
Tétralogie de Fallot
La tétralogie de Fallot décrite pour la première en 1988 est la cardiopathie congénitale cyanogène la plus fréquente. Elle est définie par l’association de quatre anomalies :
– CIV sous-pulmonaire ;
– aorte à cheval au-dessus de la CIV ;
– sténose pulmonaire (infundibulaire, valvulaire ou supra-valvulaire) ;
– hypertrophie du ventricule droit.
La recherche d’anomalies d’implantation coronaire est également primordiale.
La cardiopathie peut être associée à des syndromes génétiques, notamment la microdélétion 22q11.
La cyanose rend obligatoire la réparation chirurgicale qui a lieu dans l’enfance (normalement dans les premiers mois de vie) consistant en une fermeture de la CIV par un patch, une résection du muscle infundibulaire et un patch d’élargissement de la voie pulmonaire, amenant à souvent sacrifier la valve pulmonaire.
Il est donc essentiel de détecter les complications arrivant à l’âge adulte, secondaire à cette réparation, dépistées par l’échographie cardiaque :
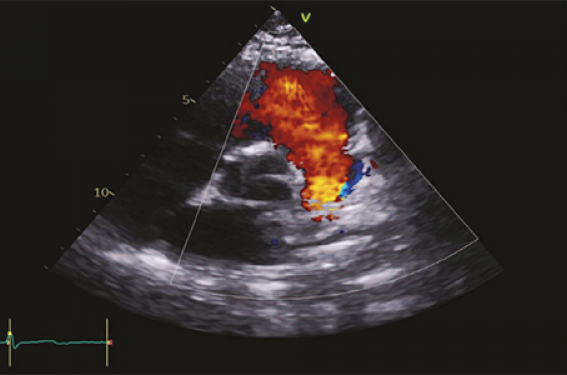
– fuite pulmonaire libre pouvant entraîner à long terme une dilatation et une dysfonction du ventricule droit (figure 5) ;
– sténose résiduelle de la voie pulmonaire ;
– CIV résiduelle sur déhiscence ou rupture du patch ;
– dilatation progressive de l’aorte avec fuite aortique sévère ;
– dysfonction du ventricule gauche dont le mécanisme est multiple (cyanose chronique, interdépendance interventriculaire, anomalies coronaires…) ;
– les troubles du rythme supraventriculaires, ventriculaires et le risque de mort subite, liés aux cicatrices de la chirurgie.
Figure 5. Tétralogie de Fallot opérée avec fuite pulmonaire libre. Échographie cardiaque en petit axe montrant une fuite libre. Intérêt de l’IRM cœur 4D flow (à droite) pour quantification de la fuite et évaluation des volumes ventriculaires.
Le risque d’endocardite infectieuse, en particulier si présence d’une prothèse valvulaire.
Toutes ces complications évolutives potentiellement mortelles ou réduisant la qualité de vie de ces patients amènent à une évaluation hémodynamique et/ou rythmique complète par un centre expert en cardiopathie congénitale de l’adulte avec une prise en charge multimodale (échographie cardiaque, imageries en coupe avec TDM/IRM-4D cardiaque, épreuve d’effort avec VO2, cathétérisme cardiaque congénital, stratification rythmique avec exploration électrophysiologique…), et multidisciplinaire en vue d’une éventuelle réinterventions/réopérations (essentiellement valvulation pulmonaire chirurgicale ou percutanée dont les indications reposent sur des critères composites : symptômes, dilatation ventriculaire droite sur l’IRM cardiaque et dysfonction ventriculaire droite).
Quelques notions des cardiopathies congénitales sévères
Qu’appelle-t-on un ventricule droit systémique ?
Il s’agit d’un ventricule droit anatomique (avec ses trabeculations importantes), placé sous la valve aortique et l’aorte, donc en position dite systémique. C’est le cas dans deux situations : les doubles discordances (ou transposition des gros vaisseaux corrigée) ou une double inversion native des connexions atrio-ventriculaires et ventriculo-artérielle.
Les transpositions des gros vaisseaux opérées par un switch atrial (opération de Seening ou Mustard).
À terme, ce ventricule droit et sa valve atrio-ventriculaire tricuspide, qui n’est pas « équipée » pour supporter une circulation systémique, évoluent vers une dilatation et une dysfonction, souvent concomitante d’une fuite de la valve tricuspide. En échocardiographie, on peut reconnaître ce ventricule droit lorsque sa valve atrio-ventriculaire (la valve tricuspide) est plus « apicalisée » que la valve controlatérale (la valve mitrale), qui est connectée à l’aorte.
Il est difficile de décider en particulier dans les doubles discordances du timing du remplacement valvulaire tricuspide éventuel, et des modes de stimulation ventriculaire en cas de trouble conductif.
Cœur univentriculaire et circulation de Fontan ?
Il s’agit de plusieurs entités anatomiques qui, in fine, conduisent à l’existence d’un seul ventricule fonctionnel, entraînant un état de cyanose, car les retours veineux systémiques et pulmonaires se mélangent dans une cavité ventriculaire unique ou principale.
La chirurgie qui est palliative (circulation de Fontan) consiste à transformer ce circuit « mélangé » en un circuit en série (en séparant le circuit de retour veineux, sans pompe ventriculaire « anastomoses cavo-pulmonaires » et la cavité ventriculaire vers l’aorte).
Les complications liées à cette physiologie sont multisystémiques (affectant tous les organes, dont le rein, le foie, le tube digestif…) et nécessitent un suivi exclusivement dans un centre expert en cardiologie congénitale de l’adulte.
Syndrome d’Eisenmenger, quèsaco ?
Le syndrome d’Eisenmenger est défini par une hypertension artérielle pulmonaire (HTAP) fixée secondaire à un shunt gauchedroit initial (CIV, CIA, CAV ou canal artériel persistant), dont le shunt s’est secondairement inversé (droit-gauche), responsable d’une cyanose chronique. À ce stade Il ne faut jamais fermer ce shunt, car il sert de soupape au ventricule droit qui se retrouverait en face de résistances dites suprasystémiques (supérieures à celle de l’aorte).
Les patients atteints de ce syndrome doivent également être adressés à un centre d’expertise pour initier un traitement vasodilatateur pulmonaire.
Comment vivre avec une cardiopathie congénitale en quelques mots
Pratique sportive
L’évaluation sportive doit tenir compte de la cardiopathie, de son évolution, du type de sport et du niveau d’effort. Globalement, les complications durant l’exercice physique, y compris la mort subite chez les patients avec une cardiopathie congénitale sont rares.
La pratique d’une activité physique modérée régulière est recommandée pour la plupart des patients avec une cardiopathie congénitale.
En revanche, les patients avec une dysfonction du ventricule systémique, un obstacle gauche, une hypertension pulmonaire, un terrain arythmogène ou une dilatation aortique, doivent être évalués et la pratique d’une activité physique de faible intensité peut être recommandée.
Grossesse et contraception
Avant une grossesse, toute patiente avec une cardiopathie congénitale (en particulier celles de complexité moyenne à sévère) doit bénéficier d’une évaluation spécialisée par une équipe multidisciplinaire incluant un obstétricien, afin d’organiser la prise en charge anténatale et postnatale (types de surveillance et choix de la maternité). Le risque maternel est basé sur la classification de l’OMS modifiée (tableau 2).
La contraception est aussi un sujet important pour ces patientes. La contraception oestro-progestative est presque efficace à 100 %. Cependant elle n’est pas recommandée chez les patientes à haut risque thromboembolique (cyanose, circulation de Fontan, valves mécaniques, antécédents de thrombose, hypertension pulmonaire). D’autres méthodes de contraception (notamment progestatives seules) doivent alors être discutées avec le cardiologue congénitaliste et le gynécologue.
Conduite à tenir devant une « urgence congénitale »
Toute syncope chez un patient avec une cardiopathie congénitale doit être admise dans une unité de surveillance continue. Chaque cardiopathie congénitale possède un risque intrinsèque soit de troubles de conduction, soit de troubles de rythmes supra-ventriculaires et/ou ventriculaire ; tous deux pouvant être à l’origine de mort subite (tableau 3).
Tout trouble de rythme, y compris supra-ventriculaire, y compris asymptomatique, chez un patient avec une cardiopathie congénitale de complexité moyenne à sévère, ne doit pas être banalisé et doit être systématiquement hospitalisé dans un centre expert, pour une stratégie de réduction rapide (médicamenteuse, électrique ou ablation). Le risque de défaillance cardiaque et de mort subite est majeur, à très court terme.
Toute fièvre chez un patient avec une cardiopathie congénitale, quelle que soit la complexité, est une endocardite infectieuse jusqu’à preuve du contraire, potentiellement mortelle, et doit être investiguée dans un centre médico-chirurgical.
Tout signe d’insuffisance cardiaque de novo chez un patient avec une cardiopathie congénitale doit être hospitalisé dans un centre spécialisé en cardiopathie congénitale de l’adulte. L’insuffisance cardiaque est en effet un problème fréquent et surtout la première cause de mortalité dans cette population.
En pratique
▸ La population des adultes avec une cardiopathie congénitale grandit et dépasse la population pédiatrique.
▸ La prise en charge diagnostique et thérapeutique qui est multimodale et multidisciplinaire doit se faire dans un centre d’expertise en cardiologie congénitale adulte. Le cardiologue travaillant en ville ou à l’hôpital a un rôle clé dans l’orientation des patients avec une cardiopathie congénitale, vers un centre d’expertise.
▸ Le suivi doit être réalisé par un cardiologue congénital pour les cardiopathies congénitales de complexité sévère, et peut être organisé en alternance avec un service de cardiologie conventionnelle pour les cardiopathies congénitales de complexité moins sévères.
▸ L’échographie cardiaque est l’outil clé pour le diagnostic et le dépistage des complications. Très souvent, une imagerie en coupe est nécessaire.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
