

Publié le 13 oct 2015Lecture 9 min
Postconditionnement : protéger le myocarde ischémique à la reperfusion
M. COUR, L. ARGAUD, Inserm U886 Cardioprotection, service de réanimation médicale ; groupement hospitalier Edouard-Herriot, Lyon, J. LOUFOUAT, M. OVIZE, Inserm U886 Cardioprotection, Lyon
L'infarctus du myocarde (IDM) demeure la première cause de décès dans le monde, alors même que le développement des unités de soins intensifs et des procédures de reperfusion a considérablement réduit sa mortalité précoce. Les conséquences de l'IDM en termes de morbi-mortalité et de pronostic fonctionnel sont directement liées à la taille de l'infarctus, qui est certes due à la durée de l'occlusion coronaire, mais aussi paradoxalement à l'importance des lésions myocardiques consécutives à la reperfusion. Des interventions thérapeutiques adjuvantes de la reperfusion coronaire, connues sous le nom de postconditionnement, sont aujourd'hui disponibles au laboratoire pour protéger le myocarde des conséquences de la reperfusion. Les premières applications cliniques de ces travaux offrent de réelles perspectives d'amélioration de la prise en charge des patients à la phase aiguë de l'IDM. Et au-delà, l'ensemble des pathologies cardiovasculaires mettant en jeu un mécanisme physiopathologique d'ischémie-reperfusion pourrait bénéficier à terme de cette recherche translationnelle.
Nécrose de reperfusion
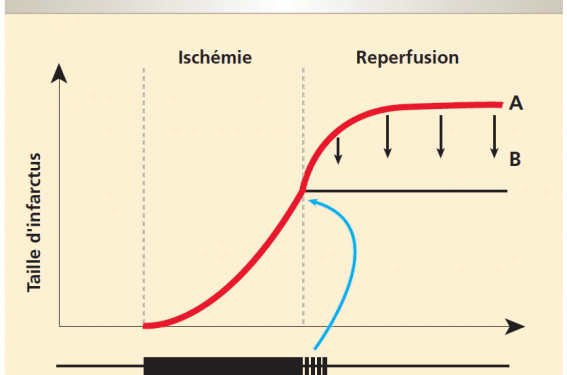
Jusqu’à récemment, la reperfusion n’était envisagée que comme un phénomène bénéfique capable de « sauver » le myocarde ischémique et ainsi stabiliser la taille de l’infarctus à un niveau déterminé par la fin de l’ischémie. Nous savons aujourd’hui que la reperfusion est, par elle-même, à l’origine d’une extension des lésions cellulaires (figure 1). La preuve de cette « nécrose de reperfusion », qui pourrait compter pour 50 % de la taille finale de l’infarctus, est apportée expérimentalement par le fait que des interventions qui n’ont lieu qu’au moment de la désocclusion coronaire sont capables de réduire considérablement la taille de l’IDM(1).
Les dégâts myocardiques contemporains de la recirculation coronaire correspondent à des mécanismes physiopathologiques complexes, dépendant, bien sûr, de la période ischémique, mais initiés lors des toutes premières minutes de reperfusion faisant suite à une occlusion coronaire prolongée. En effet, dès le début de la reperfusion, au moins trois mécanismes délétères apparaissent :
1. Un stress radicalaire intense correspondant à une production excessive de radicaux libres de l’oxygène par une chaîne respiratoire mitochondriale lésée par l’ischémie et brutalement soumise à une réoxygénation.
2. Une surcharge calcique cytosolique puis mitochondriale consécutive aux mécanismes compensateurs de l’excès de protons.
3. Une rapide correction de cette acidose, avec même une tendance transitoire à l’alcalose intracellulaire(2).
Figure 1. Au cours de l’ischémie-reperfusion myocardique, une partie des cardiomyocytes est irréversiblement lésée par une période d’occlusion coronaire prolongée. Néanmoins une part importante de la taille de l’infarctus est due à des lésions initiées à la reperfusion, touchant du myocarde encore viable à la fin de l’ischémie (A). Cette « nécrose de reperfusion » peut être considérablement limitée (B), jusqu’à 50 % de la taille finale de l’infarctus, par une intervention de type postconditionnement (PostC).
Ces événements conjugués altèrent le fonctionnement mitochondrial, avec en particulier une perte de l’intégrité de la membrane mitochondriale interne, indispensable au maintien d’un potentiel de membrane dont dépend la production d’ATP(3).
Cette perméabilisation de la membrane interne de la mitochondrie correspond en réalité à l’ouverture d’un méga-canal fonctionnel, appelé pore de transition de perméabilité (PTP), capable de faire communiquer largement la matrice mitochondriale et le cytosol, à l’origine d’un gonflement matriciel et de l’effondrement des capacités de phosphorylation oxydative, par découplage de la chaîne respiratoire. La structure du PTP est aujourd’hui incomplètement connue, même s’il s’agit probablement d’un assemblage multiprotéique des constituants naturels des membranes mitochondriales et de ceux de l’espace intermembranaire(3). La cyclophiline D est la seule protéine constitutive (appartenant à la matrice mitochondriale) connue de ce pore, ayant la particularité de fixer la ciclosporine A (CsA), qui par ce mécanisme, et indépendamment de ses effets immunosuppresseurs bien connus, est l’inhibiteur de référence du PTP(4). On comprend ainsi toute l’importance de la mitochondrie, organite indispensable à la vie des cellules eucaryotes, mais ayant aussi à la reperfusion un rôle décisif dans l’induction de la mort cellulaire. Le PTP est donc une cible thérapeutique de choix pour protéger le myocarde des conséquences de l’ischémie-reperfusion(5).
Postconditionnement : approche mécanique et pharmacologique
Le préconditionnement ischémique, qui consiste à réaliser expérimentalement des séquences courtes d’ischémie-reperfusion avant une occlusion coronaire prolongée (infarctoïde), a longtemps été considéré (depuis sa découverte en 1986) comme le gold standard en matière de cardioprotection(6). Même si les applications cliniques de ce concept expérimental étaient par nature limitées, il a néanmoins permis de pointer le rôle d’effecteur de la mitochondrie et de son PTP dans la signalisation de la protection cellulaire(7). C’est en 2003 que, par analogie avec le préconditionnement, le groupe de Vinten-Johansen aux États-Unis a décrit pour la première fois le concept de postconditioning(8). Ces auteurs ont, en effet, montré dans un modèle d’infarctus expérimental chez le chien que la réalisation de brefs épisodes d’ischémie-reperfusion (par clampage-déclampage) coronaire, juste après une occlusion coronaire prolongée, réduisait la taille de l’infarctus d’environ 50 %, soit d’une manière comparable à l’effet du préconditionnement(8). Pour être efficace, cette intervention doit impérativement être appliquée dès les toutes premières minutes de reperfusion. Ces résultats ont depuis été reproduits par de nombreuses équipes à travers le monde, dont la nôtre (figure 2)(9). Cette démonstration a confirmé de manière définitive la preuve du concept de nécrose de reperfusion(1,2).
Figure 2. Les protocoles expérimentaux de cardioprotection à la reperfusion (A) privilégient soit une approche mécanique de postconditionnement ischémique (PostC), en appliquant au début de la reperfusion des périodes brèves d’ischémie-reperfusion, comme cela est possible au cours de l’angioplastie primaire à la phase aiguë de l’infarctus du myocarde, soit une approche pharmacologique en mimant le mécanisme d’action du postconditionnement ischémique. L’inhibition du pore de transition de perméabilité (PTP) mitochondrial par la ciclosporine A (CsA), administrée dès le début de la reperfusion, induit une cardioprotection aussi puissante que celle du postconditionnement ischémique, objectivée par une réduction significative de l’aire nécrosée (AN), exprimée ici en pourcentage d’infarcissement de l’aire à risque (AR) dépendant de l’artère coronaire « coupable » (B).
* p < 0,001 versus groupe contrôle (Ctrl).
Le mécanisme d’action du postconditionnement est incomplètement élucidé. Néanmoins, le niveau de connaissance actuel laisse penser qu’il s’appuie sur les mêmes voies de signalisation cytoprotectrices ubiquitaires qu’utilise le pré- conditionnement, à savoir la voie reperfusion injury survival kinase (RISK) via l’activation de récepteurs membranaires à des substances autacoïdes (adénosine, bradykinine, dérivés opioïdes, par exemple) (figure 3)(6,10). À cela s’ajoutent probablement la modulation de la production d’espèces radicalaires de l’oxygène et le rôle du pH intracellulaire(10). La sortie plus progressive de l’acidose, due à l’application de périodes d’ischémie au début de la recirculation coronaire, contribue, en effet, à maintenir plus longtemps le PTP en configuration fermée. Quelles que soient les voies de médiation utilisées, il est clairement établi que le PTP mitochondrial a un rôle d’effecteur clé dans la transmission du signal cytoprotecteur du postconditionnement (figure 3)(9). Il est admis que l’ischémie (en particulier par l’importance de l’acidose) maintient le PTP fermé, alors que la reperfusion en conjuguant surcharge calcique, stress radicalaire et augmentation de pH intracellulaire augmente sa probabilité d’ouverture(5). Nous avons montré chez l’animal que les mitochondries de cardiomyocytes, prélevées in vivo après un IDM, présentaient une plus grande susceptibilité d’ouverture du PTP, et que ce phénomène était prévenu de la même façon par le préconditionnement ou par le post-conditionnement ischémique(9).
Figure 3. Le mécanisme d’action du postconditionnement ischémique (PostC) est médié par la voie de signalisation RISK ( Reperfusion Injury Survival Kinase) et en particulier deux cascades de médiations cytosoliques : celle de la phosphatidylinositol-3 kinase-Akt (PI3K-Akt) et celle de l’ Extracellular Signal Regulated Kinase (MEK-ERK), qui inhibent la Glycogen Synthase Kinase-3 β (GSK-3 β). Le postconditionnement prolonge également l’acidose intracellulaire pendant les premières minutes de reperfusion et module la production des radicaux libres de l’oxygène. Tous ces mécanismes concourent à inhiber l’ouverture du pore de transition de perméabilité (PTP) mitochondrial, effecteur final de la protection cellulaire, dont l’inhibiteur pharmacologique de référence est la ciclosporine A (CsA).
La meilleure connaissance de la transduction du signal cytoprotecteur a naturellement conduit à des perspectives pharmacologiques du postconditionnement s’appuyant sur ces voies de signalisation. Le groupe de D. Yellon, à Londres, a en particulier développé des approches expérimentales d’activation de la voie RISK (par exemple par des agonistes de la protéine kinase C, des facteurs de croissance comme l’insuline, l’érythropoïétine...), dont la translation en clinique n’a pas aujourd’hui fait la preuve de son efficacité(2). Nous privilégions, quant à nous, une approche thérapeutique d’inhibition du PTP à l’aide de la CsA ou de ses dérivés non immunosuppresseurs spécifiques du PTP (figure 2)(10). Nous avons montré au laboratoire l’efficacité de ces thérapeutiques sur l’ensemble des modèles animaux d’IDM dont nous disposons, avec une efficacité de la cardioprotection superposable à celle du postconditionnement ischémique (mécanique), laissant présager d’intéressantes perspectives de développement en clinique(10).
Perspectives cliniques du post-conditionnement
Les applications du postconditionnement chez l’homme sont de plus en plus évidentes. L’équipe de M. Ovize à Lyon a récemment montré que le postconditionnement méca- nique, par angioplastie coronaire, à la phase aiguë de l’IDM limite la taille de l’infarctus et améliore le pronostic fonc- tionnel des patients(11,12). Néanmoins, même si ce concept de « postconditionner » le myocarde au début de la reper- fusion par angioplastie coronaire est évidemment sédui- sant, il souffre d’au moins deux limites : le pourcentage encore important de patients traités par thrombolyse et/ou ne bénéficiant pas d’une revascularisation précoce par angioplastie, ainsi que le développement des techniques de thromboaspiration qui semblent intéressantes pour améliorer le débit sanguin myocardique et le pronostic des patients, mais incompatibles avec un postconditionnement ischémique précoce(10). Dans une étude de « preuve de concept », Piot et coll. ont également retrouvé les mêmes ésultats en termes de diminution de la taille de l’infarctus, à l’aide d’un postconditionnement pharmacologique par la CsA (figure 4)(13). L’effet de cette réduction de la taille de l’infarctus est encore visible 6 mois après l’IDM et s’accompagne entre autres d’une moindre dilatation ventriculaire gauche(14). Ces résultats prometteurs, applicables à terme à l’ensemble des patients bénéficiant d’une reperfusion coronaire, quelle qu’elle soit, devront, bien sûr, être confirmés à l’avenir par une large étude multicentrique.
Figure 4. L’administration perangioplastique de cyclosporine A (CsA) à des patients à la phase aiguë de l’infarctus du myocarde, comparativement à un groupe de patients contrôles (Ctrl), diminue (d’environ 40 %) le relargage de créatine kinase (CK) et de troponine Ic, don't l’aire sous la courbe Durant les 3 premiers jours d’évolution après désocclusion coronaire est un marqueur indirect de la taille de l’infarctus (d’après(13)).
Au-delà de l’IDM, qui reste le modèle d’étude par excellence des conséquences de l’ischémie-reperfusion, nombre de situations physiopathologiques mettent en jeu des mécanismes d’hypoxie-réoxygénation à l’échelon cellulaire. L’ubiquité des mécanismes mitochondriaux de protection cellulaire activés par le postconditionnement, quels que soient l’organe considéré (cœur, cerveau, foie, rein...) et l’espèce animale étudiée (tous les animaux de laboratoire étudiés ainsi que l’homme), que l’ischémie-reperfusion soit focale, régionale ou globale, laisse à penser que le champ d’application du postconditionnement (ischémique ou pharmacologique) ne demande qu’à être investigué(10). D’ores et déjà, que ce soit dans la problématique de conservation des greffons avant transplantation, dans l’accident vasculaire cérébral, dans l’état de choc ou dans l’arrêt cardiaque, pour ne parler que du domaine cardiovasculaire, plusieurs applications de type postconditionnement se sont avérées efficaces pour prévenir la mort cellulaire et/ou limiter les dysfonctions d’organes(10). Nous avons, par exemple, montré récemment, dans un modèle d’arrêt cardiaque chez le lapin, l’intérêt de l’administration (concomitante du début de la réanimation cardio-pulmonaire) d’un inhibiteur du PTP pour limiter le syndrome post-arrêt cardiaque (caractérisé par une défaillance multiviscérale) et in fine améliorer la survie(15).
L’ensemble de ces applications potentielles du postconditionnement doit désormais faire l’objet d’études cliniques appropriées afin de confirmer chez l’homme la réalité de ces concepts issus du laboratoire.
Ce qu’il faut retenir
La reperfusion est dans l’infarctus du myocarde une « arme à double tranchant » capable de « sauver » le myocarde ischémique, mais aussi d’induire des lésions au début de la recirculation coronaire.
La mitochondrie par l’intermédiaire d’un de ses constituants, le pore de transition de perméabilité (PTP), a un rôle dual dans la survie des cardiomyocytes.
Le postconditionnement ischémique (mécanique), qui consiste à appliquer des périodes brèves d’ischémie-reperfusion après une occlusion coronaire prolongée, est un puissant mécanisme cardioprotecteur, y compris chez l’homme via l’inhibition du PTP mitochondrial.
Le postconditionnement pharmacologique est une intervention thérapeutique médicamenteuse cytoprotectrice, mimant le mécanisme d’action du post- conditionnement ischémique.
La ciclosporine A (CsA), indépendamment de ses effets immunosuppresseurs, en inhibant le PTP, est un puissant agent de postconditionnement pharmacologique, dont l’effet est prouvé depuis peu en clinique.
L’ubiquité des phénomènes mitochondriaux de protection cellulaire au cours de l’ischémie-reperfusion (régionale ou globale) laisse entrevoir de nombreuses autres perspectives cliniques de postconditionnement pharmacologique dans le domaine cardiovasculaire.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
